
Abstract
Introduction
An increasing number of cases of comorbid hereditary spastic paraplegia (HSP) and multiple sclerosis (MS) have been described. We report a patient with the SPG3A form of HSP and features of relapsing-remitting MS (RRMS). We took this opportunity to review the current literature of co-occurring MS and HSP.
Method
The patient underwent clinical, laboratory and neuroimaging evaluations. We performed a literature search for cases of HSP and MS. The 2017 McDonalds Criteria for MS were retrospectively applied to the selected cases.
Results
A 34-year-old woman, presenting a molecular diagnosis of SPG3A, complained subacute sensory-motor symptoms. Spinal MRI disclosed T2-hyperintense lesions at C2, T6 and T4 level, the latter presenting contrast-enhancement. CSF analysis showed oligoclonal bands. She was treated with intravenous high-dose steroids, with symptom resolution. The literature review yielded 13 papers reporting 20 possible cases of MS and HSP. Nine patients (5 M, median age 34) met the 2017 McDonald criteria. Five (25%) received a diagnosis of RRMS and four (20%) of primary progressive MS. Brain MRI showed multiple WM lesions, mostly periventricular. Six of seven cases (85.7%) had spinal cord involvement. Oligoclonal bands were found in 6/8 (75%) patients. Seven patients (77.7%) improved/stabilized on immunotherapy.
Conclusion
This is the first description on the association between SPG3A type of HSP and MS. This report adds to the other reported cases of co-occurring HSPs and MS. Although it remains unclear if this association is casual or causal, clinicians should be aware that an HSP diagnosis does not always exclude a concomitant MS.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hereditary spastic paraplegia (HSP) is a group of clinically and genetically heterogeneous disorders that share the primary feature of a progressive lower extremity weakness and spasticity [1]. Multiple sclerosis (MS) is an autoimmune disease of the central nervous system characterized by inflammatory demyelination [2]. Despite differences in the underlying pathogenetic mechanisms, HSP shares with multiple sclerosis (MS), particularly its primary progressive (PPMS) variant, some clinical features, including the progressive degeneration of the corticospinal tracts. Fewer studies investigated the association between gene variants in HSP-associated genes and MS outcome [3,4,5] with variable results. Conversely, increasing cases of co-occurrence of HSP and MS, both the relapsing-remitting (RRMS) and the PPMS variants, in the same patient or in the same family, have been described [4, 6,7,8,9,10,11,12,13,14,15,16,17,18]. Here, we report a new HSP patient harbouring a pathogenic mutation in ATL1/SPG3 and presenting RRMS. We also reviewed the current literature of co-occurring MS and HSP.
Methods
We searched PubMed and Scopus for articles published up to February 2022. We used the search terms: (“hereditary spastic paraparesis”) OR (“hereditary spastic paraplegia*”) AND (“multiple sclerosis”), restricting the search to studies published in English. Bibliographies were examined manually for further eligible articles. Duplicate cases were identified whenever possible and, in overlapping cohort cases, only the larger was included. The results were screened independently by two reviewers (GR and MPG) who reached a consensus on potentially eligible articles. Extracted data were reported in a predefined form including sex, age at evaluation, gene mutation and inheritance pattern, age at onset, clinical features, neuroimaging and CSF findings, immunotherapy and outcome. The 2017 McDonalds Criteria for MS [19] were retrospectively applied independently by two reviewers (GR and MPG) to the selected cases.
Results
Case report
We describe the case of a 34-year-old woman presenting HSP due to a pathogenic mutation in the ATL1 gene (SPG3A form). Her first symptoms appeared at the age of 3 with progressive rigidity of the legs and gait impairment. The mother and the maternal grandfather, uncle and aunt complained of the same symptoms since a young age. At the age of 28, molecular analysis revealed a heterozygous c.715C>T (p.R239C) variant in ATL1 [20]. At that time, the neurological examination revealed moderate spasticity with spastic gait. A spinal MRI only disclosed a small meningioma at level C6–C7 without significant compression. Brain MRI and electromyography were normal.
During the following years, the spastic paraparesis slowly progressed.
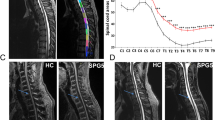
A follow-up brain MRI at the age of 34 disclosed some supratentorial small white matter changes, hyperintense on T2/FLAIR images without contrast-enhancement, some of these showing a mild diffusion restriction (Fig. 1A, B, C, D), not associated with specific symptoms. Two months later, the patient experienced sudden onset of reduced tactile sensation, tingling paraesthesia and dysesthesia, initially confined to her left foot, then spreading to the contralateral limb and ascending just below her breasts. This was associated with a worsening of the gait impairment. Spinal MRI disclosed a contrast-enhancing, T2-hyperintense lesion involving the dorsal midline part of the spinal cord, at T4 level (Fig. 1E, F, G, H). Furthermore, T2 hyperintense lesions without contrast-enhancement were observed at C2 and T6 level. CSF analysis showed mild leucocytosis (19 cells/mmc), increased IgG (10.4 mg/dl; cut-off < 3.4 mg/dl), abnormal IgG index (3.57; cut-off: 0.66) and oligoclonal bands. Infections were excluded, and autoimmunity panels were negative, including anti-AQP4 and anti-MOG antibodies. A MS diagnosis was made, and the patient was treated with intravenous high-dose steroids, with gradual symptom resolution. MRI performed after 6 months showed an improvement of the spinal lesions (Fig. 1M, N). No more attacks occurred during a 2-year follow-up.

Brain and spinal cord MRIs. In the upper row, the baseline lesion load is visible. (A, B, C): axial T2-weighted fluid attenuation inversion recovery (FLAIR) images showing little hyperintense deep white matter lesions in the left frontal lobe. D Sagittal fluid attenuation inversion recovery (FLAIR) image T2 does not show any abnormal signal in other typically involved structures (corpus callosum, pons, bulbo-medullary junction and cerebellum). In the middle row, spinal cord MRI (2 months later): E Sagittal TSE T2-weighted and F axial MERGE T2 showing C2 cervical spinal cord lesion on the right medullar side. G Sagittal TSE T2-weighed images showing two dorsal lesions at T4 and T6 levels. Swollen spinal cord is visible at T4 level. H Sagittal TSE T1-weighted after gadolinium shows pathological contrast enhancement of the most cranial dorsal lesion (T4 level). In the lower row, brain and spinal follow up MRI (8 months later). I, L Axial T2-weighted fluid attenuation inversion recovery (FLAIR) images showing unchanged small isolated white matter lesion in left frontal lobe. M, N Spinal cord images showing less swollen medulla with slight reduction of the two dorsal spinal lesions without contrast enhancement. Note the incidental finding of a small meningioma at level C6-C7 (E)
The mother of the patient, carrying the same ATL1 mutation and affected by a pure spastic paraplegia since her childhood, did not disclose white matter lesions at brain MRI.
Literature review
Overall, 13 papers were selected (Fig. 2), and 20 possible cases of MS and HSP were identified (Table 1) [6,7,8,9,10,11,12,13,14,15,16,17,18]. These included 6 (30%) cases with reported RRMS, 6 (30%) with reported PPMS, one (5%) reported as CIS, one patient with MRI criteria of dissemination in space and one without MS diagnosis. In the remaining cases, the MS course was not reported. For 8 (40%) cases, there were not enough data to achieve the diagnosis, whereas one case did not meet the criteria for MS. Nine patients (5 M, 4 F, median age 34 years, range 10–49) met the 2017 McDonald criteria for MS [19] and were included in the analysis. Five (25%) received a diagnosis of RRMS and four (20%) of PPMS. HSP-associated mutations were found in SPG4/SPAST in 3 cases, SPG2/PLP1 in further 3, SPG11/KIAA1840 in 2 and SPG7 in one individual. Brain MRI was available in all cases, showing the presence of multiple WM lesions, mostly periventricular. The administration of gadolinium was reported in 5 cases and contrast enhancement demonstrated in 4 (80%). Spinal cord involvement was assessed in 7 cases, demonstrating the presence of single or multiple lesions in 6 (85.7%). Oligoclonal band presence was reported in 6 (75%) of the 8 cases for which CSF analysis was available. All cases underwent immune treatment, including iv methylprednisolone (IVMP) during the acute phase in 8 cases and disease-modifying therapies (DMTs), such as interferon 1 beta, teriflunomide (TFN), dimethyl fumarate (DMF), natalizumab (NTZ) or rituximab (RTX) in 5 cases. Seven patients (77.7%) improved or stabilized on immunotherapy, whereas two, treated only with steroids, showed no response.

Flow diagram. Study selection flow chart
Discussion
Our patient presented the classic SPG3A phenotype, with a pure early-onset HSP. SPG3A is caused by mutations in ATL1 gene on chromosome 14. ATL1 codes for atlastin, a GTPase protein predominantly expressed in pyramidal neurons and grouped in the dynamin family, involved in many cellular processes, as cytoskeletal function, mitochondrial maintenance and in synaptic vesicle recycling [20]. At the age of 34, the patient experienced a reversible sensory deficit recognized as a MS attack. This diagnosis was supported by the neuroimaging and laboratory findings and further corroborated by the response to IVMP.
A casual association can be possible, although, based on a prevalence of 176 × 10−5 for MS in Italy [21] and of 1.8 × 10−5 for autosomal dominant HSP [22], the association would have a probability of 0.3 × 10−7. The increasing number of reports in which HSP and MS occur in the same patient or family [4, 6,7,8,9,10,11,12,13,14,15,16,17,18], can suggest that this association is more than the result of a chance.
From our literature review, we identified 20 patients with the possible association of MS and HSP [6,7,8,9,10,11,12,13,14,15,16,17,18], mostly related to SPAST mutations. However, in many cases, there were not enough data to achieve a definite MS diagnosis according to the 2017 McDonald criteria for MS [19]. In other cases [15, 16], the same authors pointed out a MS misdiagnosis due to a mimicry between the two conditions, although one case still met the 2017 McDonalds criteria for MS [16]. A similar misdiagnosis cannot be excluded also in other cases. Indeed, the 2017 McDonalds criteria are highly sensitive but not specific for MS, and misdiagnosis is frequent [23]. Diffused WM hyperintensities on T2-weighted images have been reported in patients with HSP associated with different mutations including genes associated with the SPG11, SPG2, SPG5, and SPG35 subtypes of HSP [24], in some cases leading to a misdiagnosis of MS [15, 16]. Similarly, another reported case with SPG4 HSP met the dissemination in space criteria at brain MRI, although the atypical morphology/distribution of lesions was red flags for an alternative diagnosis [11]. However, such lesions have never been reported in patients with SPG3A [25]. Moreover, oligoclonal bands have been identified also in subjects with Mendelian and mitochondrial neurological disorders [26], and therefore, their presence is not highly specific for MS [27]. However, spinal cord inflammatory lesions were confirmed by MRI in 85% of assessed cases, a finding not reported in HSP [24]. Moreover, 80% of cases with reported MS and HSP undergoing immunotherapy, including ours, presented some degree of clinical improvement or disease stabilization, supporting the presence of a concomitant ongoing inflammatory process.
It has been demonstrated that patients with PPMS and SPMS are enriched for HSP-related mutations causing axonal injury, which could contribute to disease evolution in progressive MS [4, 5]. On the other hand, a broader link between inflammation and neurodegenerative diseases has been described [28]. According to this, misfolded proteins or nucleic acids in non-physiological compartments act as damage-associated molecular patterns (DAMPs), activating microglia and other cell types that express protein recognition receptor (PPR). This eventually triggers inflammatory events that contribute to neuronal dysfunction and death. A similar process could also take place in HSPs, where chronic neuronal damage releases antigens, possibly triggering an autoimmune response. Notably, MS has been described in association with other genetic diseases (e.g. neurofibromatosis and LHON) [29, 30]. This would be in line with the “intrinsic model” of MS pathogenesis, which hypothesises that the initial event that triggers autoimmunity happens inside the CNS and leads to release of antigens peripherally.
In conclusion, our case adds to the literature supporting the need to further investigate a possible pathogenic link between HSP and MS. Furthermore, these cases offer further food for thought, that is, in a patient suffering from hereditary spastic paraplegia or other neurodegenerative disease, one must not make the mistake of underestimating the appearance of new or atypical symptoms.
References
Mackay-Sim A (2021) Hereditary spastic paraplegia: from genes, cells and networks to novel pathways for drug discovery. Brain Sci 11:403
McGinley MP, Goldschmidt CH, Rae-Grant AD (2021) Diagnosis and treatment of multiple sclerosis: a review. JAMA 325:765
DeLuca GC, Ramagopalan SV, Cader MZ et al (2007) The role of hereditary spastic paraplegia related genes in multiple sclerosis: a study of disease susceptibility and clinical outcome. J Neurol 254:1221–1226
Criscuolo C, Carbone R, Lieto M, Peluso S, Guacci A, Filla A, Quarantelli M, Lanzillo R, Brescia Morra V, De Michele G (2016) SPG5 and multiple sclerosis: clinical and genetic overlap? Acta Neurol Scand 133:410–414
Jia X, Madireddy L, Caillier S et al (2018) Genome sequencing uncovers phenocopies in primary progressive multiple sclerosis. Ann Neurol 84:51–63
Mead SH (2001) A large family with hereditary spastic paraparesis due to a frame shift mutation of the spastin (SPG4) gene: association with multiple sclerosis in two affected siblings and epilepsy in other affected family members. J Neurol Neurosurg Psychiatry 71:788–791
Yazıcı I, Yıldırım N, Zorlu Y (2013) The coexistence of multiple sclerosis and hereditary spastic paraparesis in a patient. Neurol Int 5:6
Boucher JJ, Counihan TJ (2020) Co-incident primary progressive multiple sclerosis and hereditary spastic paraplegia (SPG4) – a case report. Mult Scler Relat Disord 44:102375
Bellinvia A, Pastò L, Niccolai C et al (2020) A new paraplegin mutation in a patient with primary progressive multiple sclerosis. Mult Scler Relat Disord 44:102302
Laurencin C, Rascle L, Cotton F, Grosset-Janin C, Bernard E, Depienne C, Vukusic S, Thobois S (2016) A rare case of SPG11 mutation with multiple sclerosis. Rev Neurol (Paris) 172:389–391
Maggi P, Absinta M, Sati P et al (2020) The “central vein sign” in patients with diagnostic “red flags” for multiple sclerosis: a prospective multicenter 3T study. Mult Scler J 26:421–432
Dürr A, Davoine C-S, Paternotte C et al (1996) Phenotype of autosomal dominant spastic paraplegia linked to chromosome 2. Brain 119:1487–1496
Rubegni A, Battisti C, Tessa A, Cerase A, Doccini S, Malandrini A, Santorelli FM, Federico A (2017) SPG2 mimicking multiple sclerosis in a family identified using next generation sequencing. J Neurol Sci 375:198–202
Warshawsky I, Rudick RA, Staugaitis SM, Natowicz MR (2005) Primary progressive multiple sclerosis as a phenotype of a PLP1 gene mutation. Ann Neurol 58:470–473
Romagnolo A, Masera S, Mattioda A, Superti G, Santorelli FM, Mongini T, Pinessi L, Cavalla P (2014) Atypical hereditary spastic paraplegia mimicking multiple sclerosis associated with a novel SPG11 mutation. Eur J Neurol 21:e14–e15
Mukai M, Koh K, Ohnuki Y, Nagata E, Takiyama Y, Takizawa S (2018) Novel SPG11 mutations in a patient with symptoms mimicking multiple sclerosis. Intern Med 57:3183–3186
Gorman MP, Golomb MR, Walsh LE, Hobson GM, Garbern JY, Kinkel RP, Darras BT, Urion DK, Eksioglu YZ (2007) Steroid-responsive neurologic relapses in a child with a proteolipid protein-1 mutation. Neurology 68:1305–1307
Varghaei P, Estiar MA, Ashtiani S et al (2021) Genetic, structural and clinical analysis of spastic paraplegia 4. https://doi.org/10.1101/2021.07.20.21259482
Thompson AJ, Banwell BL, Barkhof F et al (2018) Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 17:162–173
Zhao X, Alvarado D, Rainier S et al (2001) Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat Genet 29:326–331
Battaglia MA, Bezzini D (2017) Estimated prevalence of multiple sclerosis in Italy in 2015. Neurol Sci 38:473–479
Ruano L, Melo C, Silva MC, Coutinho P (2014) The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 42:174–183
Solomon AJ, Naismith RT, Cross AH (2019) Misdiagnosis of multiple sclerosis. Neurology 92:26–33
da Graça FF, de Rezende TJR, Vasconcellos LFR, Pedroso JL, Barsottini OGP, França MC (2019) Neuroimaging in hereditary spastic paraplegias: current use and future perspectives. Front Neurol 9:1117
Servelhere KR, Casseb RF, de Lima FD, Rezende TJR, Ramalho LP, França MC (2021) Spinal cord gray and white matter damage in different hereditary spastic paraplegia subtypes. Am J Neuroradiol 42:610–615
Weisfeld-Adams JD, Katz Sand IB, Honce JM, Lublin FD (2015) Differential diagnosis of Mendelian and mitochondrial disorders in patients with suspected multiple sclerosis. Brain 138:517–539
Intrathecal oligoclonal IgG synthesis in multiple sclerosis - Journal of Neuroimmunology. https://www.jni-journal.com/article/S0165-5728(13)00175-6/fulltext. Accessed 11 Feb 2022
Heneka MT, Kummer MP, Latz E (2014) Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14:463–477
Bergqvist C, Hemery F, Ferkal S, Wolkenstein P (2020) Neurofibromatosis I and multiple sclerosis. Orphanet J Rare Dis 15:186
Palace J (2009) Multiple sclerosis associated with Leber’s Hereditary Optic Neuropathy. J Neurol Sci 286:24–27
Acknowledgements
We thank the patient and her family for participating in this study.
Author information
Authors and Affiliations
Contributions
(1) Research project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the first draft, B. Review and Critique.
MPG: 1C, 3A, B; EM: 1C, 3A, B; FB: 1C, 3B; CT: 1B, 3B; FMS: 2C, 3B; RL: 1B, 3B; GR: 1A, 1B, 1C, 3B
Corresponding author
Ethics declarations
Conflict of interest
Liguori R: consultation fees (Alfasigma, Amicus Therapeutics s.r.l.), lecture fees (SIMG Service, Adnkronos Salute unipersonale s.r.l., Fondazione Società Italiana di Neurologia, LT3 s.r.l., First Class s.r.l.), advisory board fees (Argon Healthcare s.r.l., Editree Eventi s.r.l., PREX s.r.l., LT3 s.r.l.), congress chair fees (DOC Congress s.r.l.), and scientific meeting organization chair fees (First Class s.r.l., I & C s.r.l.).
Other authors: No disclosures.
Ethics approval
This was a retrospective case report and did not require IRB approval. Written declaration of patient consent was obtained from the patient and family. We confirm that we have read the journal’s position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Giannoccaro, M.P., Matteo, E., Bartiromo, F. et al. Multiple sclerosis in patients with hereditary spastic paraplegia: a case report and systematic review. Neurol Sci 43, 5501–5511 (2022). https://doi.org/10.1007/s10072-022-06145-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-022-06145-1