
Abstract
Noise as an environmental stressor becomes of increasing importance in our industrialized world, and especially traffic noise from the environment represents a potential novel neurodegenerative risk factor, as well as for hearing loss. A significant number of studies have been suggested that the overproduction of reactive oxygen species (ROS) has a complex role in stimulation of pathologic events. Experimental studies upon molecular pathways of traffic noise exposure proposed that it increased the level of stress hormones and mediated the inflammatory and oxidative stress (OS) pathways resulting in endothelial and neuronal dysfunction. Studies have shown that neurons are especially sensitive to OS due to high polyunsaturated fatty acids content in membranes, high oxygen uptake, and weak antioxidant defense. However, OS induces the necrotic and apoptotic cell deaths in the cochlea. Chronic noise is one of the many overall reasons of obtained sensorineural hearing loss which destroys cognitive functions in human and animals, as well as suppresses neurogenesis in the hippocampus. Nevertheless, behavioral disorders caused by noise are mainly accompanied with oxidative stress, but the clear molecular mechanism of neurodegeneration due to disruption of the pro- and antioxidant systems is still not fully understood. This paper aims to highlight the down-stream pathophysiology of noise-induced mental disorders, including hearing loss, annoyance, anxiety, depression, memory loss, and Alzheimer’s disease, describing the underlying mechanisms of induction of inflammation and oxidative stress.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sound is considered to be one of the most important means of communication, but its longevity and high level known as noise are unwanted and harmful to humans and animals. Still in the beginning of the twentieth century, Nobel Prize winner Robert Koch predicted “One day man will have to fight noise as strongly as cholera and the plague” [1]. Urbanization and economic growth are some of risk factors for environmental noise-induced health disorders. Along with intensive industrialization and modernization, noise pollution in the workplace and industry has become an unavoidable public health problem [2]. According to the World Health Organization (WHO), environmental noise influence is unfavorable for health as the consequences are the worst. It has been confirmed that exposure to traffic-related noise accounts for a yearly loss of more than 1.5 million years of healthy life with 61,000 years for ischemic heart disease, 45,000 years for cognitive impairment of children, 903,000 years for sleep disturbance, 22,000 years for tinnitus, and 654,000 years for annoyance [3].
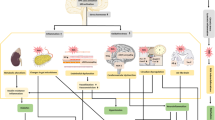
The decibel (dB) scale is a unit of measurement for noise, in which zero is counted the quietest, while sound intensity is calculated by doubling every three decibels. The sound level measured in dB (A) is to evaluate the frequencies of different sound waves by humans, which is very important for health protection. According to advisory guidelines, the risky threshold for humans’ health is 50 to 55 dB (A) [4]. Epidemiological studies have shown that level of noise may be very important in different disorders and reported that noise intensity of <60 dB may cause depressive symptoms, while >60 dB may lead to more impacts [5]. News reports document intermittent exposure to loud outdoor noise levels of 85 to 130 dB or greater is common, especially among the youngs. Noise becomes stressful at 90 dB and contributes to the development [6] of many harmful auditory and non-auditory disorders (Fig. 1).

Auditory and non-auditory effects of noise on health [11]
Chronic auditory stress leads to annoyance and sleep disturbance followed by reduction in social contacts [7]. Thereby, loud noise as a strong stressor changes the functions of the central nervous system (CNS) by breaking regulation of behavioral responses. One of the immediate targets of loud noise in the brain is the CNS, which sends stressful stimuli through the brain, altering retinal formation activity and, as a result, disrupting the sleep-waking cycle [8]. In addition, loud noise causes neuropsychiatric effects such as emotional stress and anxiety [9]. Hjortebjerg et al. have shown that road traffic resulted in attention deficit hyperactivity disorder in children [10].
Nowadays, many studies demonstrated hazardous effects of noise stress on brain functions, such as memory and learning [11, 12]. Besides, chronic noise at the level of 90–100 dB promotes development of OS in various regions of the brain followed by release of stress hormones and anxiety, i.e., the fight/flight response [13, 14]. Environmental noise exposure has been established as a phenomenon causing annoyance and mental stress reactions, resulting in sympathetic and endocrine hyperactivation, and psychological disorders such as depression and anxiety. Noise stress was found to change the brain neurotransmitter levels, cause atrophy of dendrites, and elevate plasma corticosterone levels exerting psychological symptoms. Environmental exposures cause the morpho-functional and metabolic changes resulting in diseases development, both auditory and non-auditory [15]. The non-auditory health impacts of noise are less studied, but are of crucial importance due to a wide prevalence among the millions of people. This paper aims to highlight the down-stream pathophysiology of noise-induced mental disorders, including hearing loss, annoyance, anxiety, depression, memory loss, Alzheimer’s disease, describing the underlying mechanisms which are based on induction of inflammation and oxidative stress.
Molecular mechanisms of noise-induced annoyance, anxiety, and depression and their effects
Biological systems tend to get adapted to noise influence, which depends on the person’s age, neuro-humoral state, and duration and level of noise. It is known that there are two main pathways for development of pathologies caused by noise influence, the direct and indirect ones [16]. The direct pathway is determined by the immediate interaction of the acoustic nerve with brain different structures. Annoyance, which modifies the negative emotional reactions, is a result of indirect pathway of noise exposure and conditioned by the cognitive perception of the sound, its cortical activation, and further emotional influence. The fight/flight and defeat reactions involve the hypothalamus, and thereby the autonomic nervous, the endocrine, and the limbic systems. If experienced chronically, stress-induced annoyance may even cause the development of cardiovascular disease [17]. The most common public reaction to environmental noise impact is annoyance. Severe annoyance has been associated with reduced well-being and health, and because of the high prevalence level, annoyance contributes to the exaggeration of disease course due to environmental noise. According to research data, only 20.7% of people exposed to noise were not annoyed; but 52.8% had moderate annoyance. The study results also revealed annoyance by noise exposure with depression and anxiety combination [18]. While there is a clear relationship between the objective measurement of noise and annoyance, individual properties such as noise sensitivity, also the genetic, physiological, psychological, lifestyle that increase individuals’ reactivity to noise in general, are quite important. The burden of mental disorders is quite high and disabling, ensuring the strongest impact with disability and reduced quality of life. Most contemporary etiological theories notify that stress can initiate cognitive disfunctions via increasing the risk for depression and anxiety. Regardless of the level of noise, high annoyance is followed by more mental and physical symptoms [19]. Among the possible etiological factors, the aircraft noise (affect almost 60% of the population) has been found more annoying and with stronger effects on sleep, when compared with the road and railway noise. Epidemiological data found ambient noise annoyance to be negatively associated with indicators of both physical and mental health [20].
In biological systems, anxiety is a type of emotion,which is manifested by the mental, physical, behavioral changes, resulting in neurological impairments. These pathological changes are accompanied by a fight/flight response [21]. Chronic noise stress leads to deterioration of the neuropsychiatric effects, such as anxiety and depression. Epidemiological data have shown that anxiety is one of the global public health problems with a prevalence of 14.0% (per year) impacting over 260 million people worldwide. Chronic noise as a risk factor of human health is associated with numerous health problems, such as ischemic heart disease [3], adverse birth outcomes, hearing loss, and tinnitus (Fig. 2) [22].

Molecular mechanism of anxiety under the noise influence. Under influence of noise, the brain sends messages to the autonomic nervous system, where fight/flight response is mediated by sympathetic nervous system activation. Noise stress elevated catecholamines release from suprarenals leading to increases heart rate, sweating, rate and depth of breathing, fear, and panic, as well as pain in the chest
As an environmental stressor, chronic noise can be a cause of overall psychiatric disorders, including depression, which has been considered the main prevailing mental pathology leading to disability and suicide rates. Some clinical studies have shown that high aircraft noise can be cause of depression in local residents [23].
It has been shown that chronic stress was associated with macroscopic changes in certain brain areas, resulting in the physical modifications of neuronal networks. According to some studies, stress-related effects in the prefrontal cortex (PFC) and limbic system were characterized by volume reductions of some structures and changes in neuronal plasticity due to the dendritic atrophy and decreased spine density which indicated that depressive disorders were often associated with chronic stress in humans. The basal ganglia atrophy and the gray matter significant reduction in certain areas of the PFC in the people afflicted with long-term occupational stress were observed. These alterations in a brain region can in turn cause cognitive, emotional, and behavioral dysfunctions and increase aptness to psychiatric disorders [24]. Chronic stress stimulates the production of the molecule REDD1 in the PFC via hyperactivation of the HPA axis, which then inhibits the kinase mTORC1, thus altering the phosphorylation state and function of its targets. In the brain, the interference of REDD1 with mTORC1 signaling ultimately disrupts neuronal protein and neurotrophic factors synthesis, spine formation, and synaptic plasticity. The inhibition of mTORC is crucial for synaptic impairment and appears to be a central endpoint of molecular pathways triggered by chronic stress (Fig. 3) [25].

Molecular mechanism of depression under the chronic noise conditions. Cortical centers in the brain sense a disturbing stimulus and respond by activating pathways that through the limbic system stimulate peripheral networks (the sympathetic-adrenal-medullary axis and the renin-angiotensin system, and later the HPA axis). Persistent high levels of glucocorticoids resulting from stress-induced hyperactivation of the HPA axis stimulate the production of the REDD1 molecule in the prefrontal cortex under the chronic stress. REDD1 is induced by a variety of stressors and inhibits the kinase mTORC1. In the brain, the interference of REDD1 with mTORC1 signaling ultimately impairs neuronal protein synthesis, spine formation, and synaptic plasticity
Molecular mechanism of noise-induced memory loss
Chronic noise stress was found to damage cognition, resulting in persistent memory impairments, psychiatric, dissociative, and post-traumatic stress disorders. It was shown that chronic aircraft and road traffic noise exposure has adverse effect on reading comprehension in school children. Stansfeld and Matheson’s literature review noticed that noise exposure increased demand on working memory, influenced on the processes of selectivity in memory and decision-making for carrying out tasks [26].
The mechanisms underlying behavioral changes diminish after noise influence is not fully defined. Noise exposure is likely to damage cognitive functions through two different but interrelated points. One is connected with the oxidative reaction caused by noise impact and associated with neuronal degeneration in many auditory nuclei, as well as in the brain regions critical for cognitive functions. The second is related to the hippocampal degradation and impaired spatial memory due to noise-induced modification of auditory input after hearing loss [27].
Furthermore, a study carried out on animals showed that chronic noise-induced oxidative stress caused the impairment of memory and reduction of the dendritic processes in the hippocampus (HIP) [28, 29]. The HIP is a region, which is involved in the formation of working and declarative memories, as well as indirectly connected to the auditory system by the fronto-medial cortex, insula, and amygdala, and directly by the auditory cortex and CA1 area. Therefore, this pathway works for long-term memory formation, but the acoustic signals are used for spatial memory formation. From this point of view, stimulation or loss of sound can be a reason of the HIP damage. Chronic noise disrupts spatial memory and elevates tau phosphorylation and oxidative detriment in the hippocampus of rats. Acute noise stress changes cell activity in the hippocampus and raises arc expression (hippocampus-dependent fear conditioning) in the HIP [30]. Learning and memory are connected with HIP, showing age-associated damage and age-related decrease in performing behavioral tasks due to morphological and functional modifications of the HIP. Consequently, learning and memory through the HIP are associated with integration of cognitive and emotional information and modulation of the hypothalamic-pituitary-adrenal (HPA) axis to psychological stress. It was postulated that changes in HIP, especially the amino acid neurotransmitters and their receptors, were the most essential mechanism of noise-induced cognitive impairment [31]. Changes of the amino acid neurotransmitters constitution in the brain are connected with the brain trauma, degenerative defects of CNS, and behavioral damage. Glutamate is the major excitatory, and GABA is the major inhibitory neurotransmitter in CNS. Since the basic characteristic of neuronal communications is integration of excitatory and inhibitory signals, the plasticity of synapses needs maintaining appropriate levels of these two properties for learning and memory. Chronic acoustic stress leads to overactivation of the HPA axis, excessive production of excitatory transmitters, and attenuation of inhibitory GABAergic control resulting in impairment of spatial learning and memory [32].
Rolls had shown that information was consistently processed through the hippocampal subregions, i.e., via trisynaptic loop [33]. Accordingly, information enters through entorhinal cortex (EC) projections to dentate gyrus (DG): from DG to the CA3 subregion, and from CA3 to the CA1 subregion with serially projections. The latter, in turn, sends projections back to entorhinal cortex to complete the trisynaptic loop. Consequently, injury of any component of the trisynaptic loop may lead to hippocampal dysfunction. The study carried out on animals showed that rats with CA3 lesions had significantly impaired working memory. According to some studies, chronic noise stress increased glucocorticoid levels leading to apoptosis in CA3 and CA1 regions in the HIP resulting in impairments in spatial working memory [33]. Behavioral studies showed that NMDA receptors in the CA3 subregion were involved in the acquisition of knowledge [32]. The NR2B subunits are structural and functional components of the NMDA receptor and have crucial role in learning and memory. Cui et al. found that by noise influence the NR2B expression was deceased in the DG, CA3, and CA1 regions because of changes in the Glu and its NMDA receptors, GABA production, leading to noise-induced neural apoptosis and cognition impairment (Fig. 4) [34].

Trisynaptic loop in the normal and chronic noise stress conditions. In the normal conditions, CA1 pyramidal neurons obtain information from subnetworks of the DG and CA3 as well as direct projections from the EC. The CA1 neurons compare new information from the EC with stored information via CA3 relevant in detection of error, mismatch, and novelty. Chronic noise elevates ROS level, thus becoming the cause of changes in the glutamate neurotransmitter system, which leads to apoptosis of CA1 and CA3 regions and affliction of learning and memory
Noise-induced hearing loss
One of the important health care problems is noise-induced hearing loss (NIHL) which is strongly connected with diminishing in cognition, memory, and attention [35]. It is considered to be the second most prevalent form of hearing loss after age-related hearing loss [36]. A comprehensive literature review on NIHL showed that people with hearing loss have increased social stress due to their inability to communicate with the surrounding world [37]. NIHL can be caused by a one-time exposure to an intense impulse sound or more frequently by a long-term constant exposure with sound pressure levels (SPLs) more than 80–85 dB. In urban areas, the noise levels have been monitored across different sections of the town/city, and ambient noise levels have been found to exceed the permissible limits [38]. Noise exposures of around 115–125 dB SPL bring to mechanical injury, although the level of noise exposure to the ear is <115 dB, and the injury is mostly metabolically driven. Circumstances that contribute to the development of oxidative stress at the molecular level are genetic and environmental factors, which is one of the well-established pathways underlying NIHL. Noise-induced stress elevated levels of ROS in outer hair cells (OHCs) [39]. Besides direct biochemical damage, ROS have also indirect effects due to lipid peroxidation in the cochlea and formation of toxic substances [40]. Under chronic noise influence, lipid peroxidation products promote apoptosis, accumulate isoprostanes as a vasoactive lipid peroxidation product thereby reducing cochlear blood flow [41]. ROS induces inflammation with production of pro-inflammatory cytokines, such as interleukin-6 (IL-6) and tumor necrosis factor a (TNF-a), which can themselves produce cochlear damage [42].
Free Ca2+ has been found to rise in cochlear HCs instantly when exposed to harmful noise. This increase is mediated by entry from the extracellular compartment through the channels such as L-type Ca2+ and P2X2 ATP-gated channels. Extracellular Ca2+ entry in turn can enhance the release of Ca2+ from intracellular stores [43] with further raise in free Ca2+. Increased level of free Ca2+ not only causes cytoplasmic ROS elevation, but also promotes apoptotic cell death independent of ROS, as well as promotes activation of intracellular signaling cascades of cell stress, such as mitogen-activated protein kinase (MAPK), having an essential role in injury to HCs [44]. Activation of stress-activated MAPK promotes cell differentiation, motility, cell survival, proliferation, and apoptotic cell death. MAPK acts in key pathways of inflammation and cellular stress, caused by various biological, chemical, and physical stress stimuli. NIHL includes a reduction in glutathione peroxidase (GSH-Px) activity leading to loss of outer hair cells [45]. Elevated Ca2+ may later mediate upregulation of glutamate excretion at the presynaptic junction promoting excitotoxicity (Fig. 5) [46].

Molecular pathways of noise-induced hearing loss
Noise-induced Alzheimer’s disease (AD)
AD is a common age-related neurodegenerative disease, which is accompanied by the progressive deterioration of behavior, cognitive performance, causing disorders in vital functions. Epidemiological studies showed that 11% of the world’s population living with AD is over 60 years of age and is predicted to rise to 22% or 115 million by 2050 [47]. Environmental factors including loud noise exposure as an important external source of stress are the risk factors that develop to this disease [48]. It has been shown that the residential noise level is 51–78.2 dB(A) and each 10 dB(A) increased noise level was associated with a 36% increase in the risk for mild cognitive disorders and a 29% for Alzheimer’s disease. The initial symptoms of AD development are manifested by unconscious behavior, gradual deterioration of memory—manifested by impaired perception of new information and speech disturbance [49]. The advanced stages of AD include profound memory loss, dyscoordinated movements, and hallucinations. The pathophysiology of AD is mostly connected with the extracellular sedimentation of amyloid beta (Aβ) plaques and the accumulation of intracellular tau neurofibrillary tangles (NFT), reactive microgliosis, and neuronal loss [50, 51]. The Amyloid Precursor Protein (APP) as a transmembrane protein is included in the growth, progress, recovery, and survival of neurons. One of APP isoforms—APP695—is common in neurons, whereas APP751 and APP770 are both in neurons and glial cells [52]. Anyway, some functions of APP depend on cleavage products of this protein, which occurs by α-, β-, and γ-secretase enzymes. Based on this, there are two APP processing pathways—non-amyloidogenic and amyloidogenic [53]. The non-amyloidogenic pathway includes APP processing by α-secretase and γ-secretase. APP has N-terminal and C-terminal ends embracing the neuronal membrane. α-secretase enzyme cleaves the C-terminal end near the cell surface to produce soluble sAPPα, which contributes to plasticity (growth of neurons) and neural stem cell proliferation and considered to be protective against the Aβ-induced toxicity [54]. sAPPα can further be cleaved by γ-secretase to produce AICD (Amyloid Precursor Protein Intra Cellular Domain) fragment for transcription and translocation regulation of neurons. Inversely, N-terminal fragment of APP (formed APPβ) is toxic because of bounding with death receptor 6 mediating axonal undercutting and causing death of neuronal cell [55]. In the amyloidogenic pathway, instead of α-secretase, APP cleavage is performed by the BACE-1enzym (β-secretase) resulting to release of sAPPβ and C-terminal fragment (β-CTF or C99). Then, the APP is cleaved by γ-secretase, which generates with different lengths chain amyloid peptides—Aβ37-43 [56]. From these fragments, Aβ42 and Aβ40 are considered to be the main Aβ species in the brain. Although Aβ40 is formed much more than Aβ42, however, Aβ42 due to the hydrophobicity of its two terminal residues shows a greater tendency to aggregation. Therefore, Aβ42 is the major component of amyloid plaques, which is neurotoxic and considered to be the main course of plaque formation and AD pathogenesis. Aβ peptides occurring by cleavage of APP from BACE-1 form aggregate and accumulate producing dense and insoluble plaque oligomers [57, 58]. The formed Aβ plaques store NMDAR, glucose, AMPAR [59], mAChRs, nAChR receptors causing damage in synaptic transmission by blocking ion channels and neurotransmitters through calcium dysregulation [60]. It was shown that formation of Aβ plaques is connected with cortical tau pathology as well [61]. Based on those mutations in APP and PS1/2 affect Aβ generation in AD, it can be suggested that tau pathogenesis may take place after Aβ deposition [62]. Aβ motivates tau hyperphosphorylation by activation of tau kinase GSK3β [63], as well as by Aβ-induced inflammation, activating some natural immune pathways and release of inflammatory cytokine, interleukin-1β (IL-1β) [64]. Astrocytes and microglia which are the most sources of cytokines in AD promote neuroinflammation being involved in anti- and pro-inflammatory processes, neuronal damage, and response of microglia to Aβ deposits. Increase in Aβ is connected with elevated level of pro-inflammatory cytokines, such as IL-1α, IL-6, TNF-α, and GM-CSF [65]. It has been shown that pathological deposition of Aβ leads to neuroinflammation in AD because of elevated level of pro-inflammatory cytokines, macrophage-induced inflammatory peptide, and colony-stimulating factor [66]. It was detected that caspase-1, which is an activator of cardinal pro-inflammatory cytokine IL-1β, is increased in the brains of AD patients leading to elevation of IL-1β in microglial cells surrounding Aβ plaques. Elevated level of IL-1β contributes to accumulation of Aβ, and then to APP processing and proteolysis [67]. Noise as an increasing ROS level factor and responsible for inflammation and elevated level of cytokines leads to AD progression. Deposition of Aβ results in microglial activation and increase in pro-inflammatory cytokines, and thus causes neuronal damage and loss [68]. It was shown that the brain was very assailable to ROS damage because of the low antioxidant capacity and the high level of polyunsaturated fatty acids [69].
Recent analysis of the brain tissue of residents from highly noisy polluted areas showed elevated in CD-68, CD-163, and HLA-DR positive cells, increased pro-inflammatory markers (interleukin-1β, IL1-β; cycloxygenase 2, COX2) and Aβ42 deposition, blood-brain-barrier injury, endothelial cell activation [70, 71], and brain damages in the prefrontal lobe [72]. Furthermore, animal studies have shown that noise influence elevated cytokine production [73] and MAP kinase signaling through JNK leading neurochemical changes, lipid peroxidation, behavior changes, and enhanced NFκβ expression [72]. Consequently, noise-induced stress damaged the hippocampus inducing changes in the size and number of neurons by neuroinflammation, as well as loss of synapse and neurons which predetermine clinical features of AD [27].
Noise exposure leads to an overactivation of the sympathetic system with increasing the catecholamines levels, which in turn activates endothelial NADPH oxidase, leading to oxidative stress [74]. It was shown that noise-induced stress produced long-term cognitive deterioration and degradation of hippocampal neurons [75]. OS as a result of imbalance between oxidants and antioxidants and one of the pathological states causes neuronal cell injury in brain leading to progression of AD. Under the normal conditions, the endogenous antioxidants and protective systems defend the organism from environmental oxidant harms. However, repeated exposure to environmental oxidants, particularly noise pollution, can result in intensification of antioxidant depletion leading to overproduction of ROS and damage of organ systems [76]. Many studies have documented that OS as an expression of ROS plays an important role in progression of neurodegenerative diseases, although complex pathogenesis remains unknown [77]. Cui et al. showed that noise exposure was able to elevate the rate of metabolism of the body by increasing the production of cytokines and ROS contributing to the development of OS [28].
Lipids are the major structural components of cell membranes and play a fundamental role in supporting brain physiological functions by involvement in neuronal differentiation, synaptogenesis, and brain development [78]. Lipids are highly represented in the myelin sheath which encompasses nerve cell axons and adjusts the capacity of a neuron to trigger action potentials encoding information. In addition, lipids inflect membrane fluidity adjusting trafficking channels, localization, and function of ion pumps, receptors, and transporters at the plasma membrane [79]. However, each factor capable to change lipid homeostasis influences on the membrane lipid rafts. The latter are cholesterol and sphingolipid-enriched microdomains [80], which contain synaptic-related proteins involved in synaptic transmission and plasticity. It has been shown that disorder in the function of lipid rafts–associated synaptic proteins could promote progression of neuropathological occasions and contribute to amyloidogenesis and protein aggregation [81]. As already mentioned above, App is one of the major pathological characteristics which is engaged in the pathogenesis of AD contributing to the aggregation of Aβ. The APP transmembrane-helix domain in which the Aβ sequence is inserted includes cholesterol-binding component that adjusts the subcellular location, trafficking, and proteolytic processing of APP [82]. In addition, because the membrane of neurons contains a high quantity of polyunsaturated fatty acids and low glutathione, they are very susceptible to the influence of free radicals. In its turn, Aβ is susceptible to the influence of free radicals promoting its aggregation because of the presence of metals (Cu, Fe, Al, Zn) capable to catalyze free radicals’ formation reactions [83]. Noise-induced stress promotes the generation of ROS with the following modification of macromolecules, such as lipoproteins and phospholipids, resulting in lipid peroxide formation [6]. Noise-induced OS activates series of apoptotic processes by production of p53, Bad, and Bax leading to lipid peroxidation, membrane damage, and neuronal death [84]. ROS-induced OS increases amount of Aβ peptides in progression of AD, which deplete Ca2+ storage in endoplasmic reticulum leading to overburden of cytosolic Ca2+. As a defense, the endogenous GSH levels decrease and the ROS content increases inside the cells [85]. Overproduction of ROS plays an important role in the accumulation and sedimentation of Aβ in AD, as well as serve as a marker of oxidative damage to proteins, DNA, RNA, and lipid peroxidation found in the brain of Alzheimer’s sufferers [86]. ROS activates protein kinase C, protein kinase A, and extracellular signal-regulated kinases2, causes hyperphosphorylation of tau, destabilizing microtubules and resulting in NFTs formation [32]. ROS can also activate protein kinase B, which its turn intercedes activation of glycogen synthase kinase 3α/β or GSK3α/β by inducing hyperphosphorylation of tau and NFTs formation, i.e., neuronal death [29]. Overactivation of antioxidative defense systems caused by the chronic noise stress elevates the rate of metabolism of the body leading to deposition of insoluble Aβ plaques, increased the size of plaque, synapse loss in the brain, and progression of AD [87].
Hearing loss can be considered a potential causal risk factor for cognitive decline, a proximity marker for incipient dementia. Alzheimer’s disease has been the major focus on epidemiological research assessing the risk of dementia associated with hearing loss. Hearing loss in middle age can account for about 10% of all cases of dementia, and it has been suggested that it has a direct potentiating effect on the development of neurodegeneration. It has been suggested that changes in auditory cognition may constitute an early warning signal of incipient dementia due to the computational demands of listening in a complex everyday acoustic environment. In support of this idea, predominantly central auditory deficits have been shown to predict CSF tau protein levels and regional atrophy profiles consistent with the pathology of Alzheimer’s disease in cross-sectional studies and the long-term development of a clinical syndrome consistent with Alzheimer’s disease [88, 89]. Griffiths et al. developed a model based on the interaction between auditory cognitive mechanisms and dementia pathology that is only relevant to AD. The model related to the interaction between brain activity due to auditory cognition and AD pathology focused on the typical progression from amnesic minimal cognitive impairment affecting the medial temporal lobe to established AD. Auditory cognition related to brain activity in the perisylvian language regions due to the speech-in-noise effort may interact with AD pathology to cause logopenic aphasia, a form of progressive aphasia. Hearing loss may also affect the mechanisms for auditory grouping and segregation in the posterior neocortex and predispose to another AD variant [90].
In clinics, to assess the extent and distribution of Alzheimer’s disease, AD pathology biomarkers are used in cerebrospinal fluid and brain imaging. Alexander et al. were focused on two alternative types of data: cognitive tests and brain anatomy studies using in vivo brain scans or post mortem dissection. Cognitive tests give a quantitative score of the severity of memory loss and other signs of cognitive decline. The second type of data includes brain scans either to measure atrophy patterns in different parts of the brain or the buildup of molecular markers that are associated with AD. The typical dementia syndrome of AD is expressed by remarkable episodic memory impairment, which is accompanied by the secondary deficits (word-finding skills, spatial cognition, and executive functions) [91]. The clinical criteria for the AD dementia syndrome include history of progressive cognitive degradation connected with decline in activities of daily living corroborated with neuropsychological testing. According to the new criteria, a diagnosis of probable AD can be made when the clinical and cognitive criteria are met and there is either documented progressive cognitive decline, abnormal biomarker(s) suggestive of AD, or evidence of proven AD autosomal dominant genetic mutation (PSEN1, PSEN2, and APP). The diagnosis of definite AD can only be made when there is histopathological confirmation from a biopsy or autopsy and the clinical criteria for AD [92].
Conclusion
In this review, the complex role of noise in production of ROS and the auditory and non-auditory effects, such as hearing loss and neurodegenerative and cognitive disorders including AD, annoyance, depression, and anxiety as well as basic mechanisms induced by noise exposure in the cochlea and the brain, were worked out.
Exposure to noise in biological systems activates the sympathetic and HPA systems, thereby increasing the stress hormones level. The latter promote the direct activation of ROS leading to OS and different adverse reactions. The milestones of noise-induced effects are the induction of endothelial dysfunction, oxidative stress, and inflammation. The chronic effects of noise include functional delays in brain maturation and impairment of neuro-behavioral competences. As a result of noise, OS has been implicated in different neurodegenerative disorders. Upon the data of several studies, ROS may be generated via different mechanisms and have significant roles in disease development. In progression of neurodegenerative disorders (e.g., AD), noise has a crucial role in dysfunction of mitochondria, which is connected with persistent OS. Response of the organism to the stress depends on defense mechanisms and psychophysiological and cognitive states. Noise-induced stress changes homeostasis of the neuroendocrine system leading to impairment of sleep, task performance, hearing, and emotional state. It was shown that age-related decline in hippocampal neurogenesis mediated the NIHL-related morphological alterations in microglial cells and the auditory pathway, as well as dystrophy in the hippocampus.
Noise, as an environmental pollutant, is a great and unrecognized stressor. Annoyance, depression, and anxiety from noise were shown to be the main pathological consequences and adverse health effects. The problem is lack of awareness of public issues regarding the significance of noise pollution, and the resolution to it is possible by finding the most feasible measures to control this environmental hazard.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Manukyan A (2020) Alfa-2 adrenoblokers decrease elevated carbonylation of erythrocytes’ membranes proteins and regulate behavioral changes induced by noise action. Life Sci 246. https://doi.org/10.1016/j.lfs.2020.117395
Heinonen-Guzejev M, Koskenvuo M, Mussalo-Rauhamaa H, Vuorinen HS, Heikkilä K, Kaprio J (2012) Noise sensitivity and multiple chemical sensitivity scales: properties in a population based epidemiological study. Noise Health 14:215
van Kempen E, Casas M, Pershagen G, Foraster M (2018) WHO Environmental Noise Guidelines for the European Region: a systematic review on environmental noise and cardiovascular and metabolic effects: a summary. Int J Environ Res Public Health 15:379
Centers for Disease Control and Prevention (2019) Loud noise can cause hearing loss
Christensen JS, Raaschou-Nielsen O, Tjønneland A, Overvad K, Nordsborg RB, Ketzel M et al (2016) Road traffic and railway noise exposures and adiposity in adults: a cross-sectional analysis of the Danish diet, cancer, and health cohort. Environ Health Perspect 124:329–335. https://doi.org/10.1289/ehp.1409052
Basner M, Babisch W, Davis A, Brink M, Clark C, Janssen S, Stansfeld S (2014) Auditory and non-auditory effects of noise on health. Lancet 383:1325–1332. https://doi.org/10.1016/S0140-6736(13)61613-X
Díaz J, Martínez-Martín P, Rodríguez-Blázquez C, Vázquez B, Forjaz MJ, Ortiz C, Carmona R, Linares C (2018) Short-term association between road traffic noise and healthcare demand generated by Parkinson’s disease in Madrid, Spain. Gaceta Sanitaria 32:553–558. https://doi.org/10.1016/j.gaceta.2017.01.005
Busceti CL, Di Pietro P, Riozzi B, Traficante A, Biagioni F, Nisticò R et al (2015) 5-HT2C serotonin receptor blockade prevents tau protein hyperphosphorylation and corrects the defect in hippocampal synaptic plasticity caused by a combination of environmental stressors in mice. Pharmacol Res 99:258–268. https://doi.org/10.1016/j.phrs.2015.06.017
Manukyan AL, Hunanyan LS, Melkonyan MM (2021) Alpha2- Adrenergic blockers restore noise-induced biochemical and cognitive disorders. J Clin Med Img Case Rep 1:1010
Hjortebjerg D, Andersen AM, Christensen JS, Ketzel M, Raaschou NO, Sunyer J et al (2016) Exposure to road traffic noise and behavioral problems in 7-year-old children: a cohort study. Environ Health Perspect 124:228–234. https://doi.org/10.1289/ehp.1409430
Manukyan AL, Grigoryan AS, Hunanyan LS, Harutyunyan HA, Manukyan MV, Mkrtchyan VS, Melkonyan MM (2020) Alfa2-adrenoblockers attenuate the elevated plasma cholesterol, anxiety levels and restore impaired spatial memory of rats under the chronic noise exposure. Sci Total Environ 740. https://doi.org/10.1016/j.scitotenv.2020.140390
Melkonyan M, Manukyan A, Hunanyan L, Grigoryan A, Harutyunyan H, Sukiasyan L, Danielyan L, Yenkoyan K (2021) Alpha2-adrenoblockers regulate development of oxidative stress and cognitive behaviour of rats under chronic acoustic stress conditions. Pharmaceuticals 14:529
Jafari Z, Kolb BE, Mohajerani MH (2018) Chronic traffc noise stress accelerates brain impairment and cognitive decline in mice. Exp Neurol 308:1–12
Manukyan AL, Grigoryan AS, Hunanyan LS, Harutyunyan HA, Manukyan V, Melkonyan MM (2020) Adrenergic alpha-2 receptor antagonists cease augmented oxidation of plasma proteins and anxiety of rats caused by chronic noise exposure. Noise Health 22:63–69. https://doi.org/10.4103/nah.NAH_31_19
Naqvi F, Haider S, Perveen T, Haleem DJ (2012) Sub-chronic exposure to noise affects locomotor activity and produces anxiogenic and depressive like behavior in rats. Pharmacol Rep 64:64–69
Schreckenberg D, Griefahn B, Meis M (2010) The associations between noise sensitivity, reported physical and mental health, perceived environmental quality, and noise annoyance. Noise & health 12:7–16. https://doi.org/10.4103/1463-1741.59995
Munzel T, Gori T, Babisch W, Basner M (2014) Cardiovascular effects of environmental noise exposure. Eur Heart J 35:829–836. https://doi.org/10.1093/eurheartj/ehu030
Beutel ME, Jünger C, Klein EM, Wild P, Lackner K, Blettner M et al (2016) Noise annoyance is associated with depression and anxiety in the general population- the contribution of aircraft noise. PLoS One 11:e0155357. https://doi.org/10.1371/journal.pone.0155357
Gilani TA, Mir MS (2021) A study on the assessment of traffic noise induced annoyance and awareness levels about the potential health effects among residents living around a noise-sensitive area. Environ Sci Pollut Res. https://doi.org/10.1007/s11356-021-15208-3
Banerjee D (2013) Road traffic noise exposure and annoyance: a cross-sectional study among adult Indian population. Noise Heal 15:342–346. https://doi.org/10.4103/1463-1741.116583
Jensen HAR, Rasmussen B, Ekholm O (2019) Neighbour noise annoyance is associated with various mental and physical health symptoms: results from a nationwide study among individuals living in multi-storey housing. BMC Public Health 19:1508. https://doi.org/10.1186/s12889-019-7893-8
European Environment Agency (2020) Environ Noise Europe
Dzhambov AM, Lercher P (2019) Road traffic noise exposure and birth outcomes: an updated systematic review and meta-analysis. Int J Environ Res Publ Health 16
Lucassen PJ, Pruessner J, Sousa N et al (2014) Neuropathology of stress. Acta Neuropathol 127:109–135
Ota KT, Liu RJ, Voleti B et al (2014) REDD1 is essential for stress induced synaptic loss and depressive behavior. Nat Med. 20:531–535
Babisch W (2002) The noise/stress concept, risk assessment and research needs. Noise & health 4:1–11
Shukla M, Mani KV (2020) Deepshikha, Shukla S, Kapoor N. Moderate noise associated oxidative stress with concomitant memory impairment, neuro-inflammation and neurodegeneration. Brain Behavior & Immunity – Health 5. https://doi.org/10.1016/j.bbih.2020.100089.
Cui B, Li K, Gai Z et al (2015) Chronic noise exposure acts cumulatively to exacerbate Alzheimer’s disease-like amyloid-β pathology and neuroinflammation in the rat hippocampus. Sci Rep 5:12943
Jafari Z, Okuma M, Karem H, Mehla J, Kolb BE, Mohajerani MH (2019) Prenatal noise stress aggravates cognitive decline and the onset and progression of beta amyloid pathology in a mouse model of Alzheimer’s disease. Neurobiol Aging 77:66–86
Zhao H, Wang L, Chen L, Zhang J, Sun W, Salvi RJ et al (2018) Temporary conductive hearing loss in early life impairs spatial memory of rats in adulthood. Brain Behav 8:e01004. https://doi.org/10.1002/brb3.1004
Luine VN, Wallace ME, Frankfurt M (2011) Age-related deficits in spatial memory and hippocampal spines in virgin, female Fischer 344 rats. Curr Gerontol Geriatr Res 316386. https://doi.org/10.1155/2011/316386
Cunha AOS, de Deus JL, Ceballos CC, Leão RM (2019) Increased hippocampal GABAergic inhibition after long-term high-intensity sound exposure. Plos one 14:e0210451. https://doi.org/10.1371/journal.pone.0210451
Rolls ET (2013) A quantitative theory of the functions of the hippocampal CA3 network in memory. Front Cell Neurosci 7:98
Manikandan S, Padma MK, Srikumar R, Jeya PN, Muthuvel A, Sheela DR (2006) Effects of chronic noise stress on spatial memory of rats in relation to neuronal dendritic alteration and free radical-imbalance in hippocampus and medial prefrontal cortex. Neurosci Lett 399:17–22
Eshraghi AA, Jung HD, Mittal R (2020) Recent advancements in gene and stem cell-based treatment modalities: potential implications in noise-induced hearing loss. The Anatomical Record 303:516–526
Konings A, Van Laer L, Van Camp G (2009) Genetic studies on noiseinduced hearing loss: a review. Ear Hear 30:151–159 https://pubmed.ncbi.nlm.nih.gov/19194285/
McDait D, Park AL, Lauer J, Agarwal P, Martinez R, Lopez L, Chadha S, Cieza A, Krug E, Toroyan T, Ambett R, Sebastian A, Balance S (2017) Global costs of unaddressed hearing loss and cost effectiveness of interventions: a WHO report. World Health Organization, Geneva ISBN 978-92-4-151204-6
Begam N, Bashar A (2020) Auditory effects and consequences of noise pollution in humans: a scoping review. Adv Treat ENT Disord 4:006–010
Kurabi A, Keithley EM, Housley GD, Ryan AF, Wong AC (2017) Cellular mechanisms of noise-induced hearing loss. Hearing Res 349:129–137. https://doi.org/10.1016/j.heares.2016.11.013
Yamashita D, Jiang HY, Schacht J, Miller JM (2004) Delayed production of free radicals following noise exposure. Brain Res 1019:201–209
Jaumann M, Dettling J, Gubelt M, Zimmermann U, Gerling A, Paquet Durand F, Feil S, Wolpert S, Franz C, Varakina K, Xiong H, Brandt N, Kuhn S, Geisler HS, Rohbock K, Ruth P, Schlossmann J, Hütter J, Sandner P et al (2012) cGMP-Prkg1 signaling and Pde5 inhibition shelter cochlear hair cells and hearing function. Nat Med 18:252e259
Tan WJ, Thorne PR, Vlajkovic SM (2016) Characterization of cochlear inflammation in mice following acute and chronic noise exposure. Histochem Cell Biol 146:219e230
Orrenius S, Zhivotovsky B, Nicotera P (2003) Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 4:552e565
Harr MW, Distelhorst CW (2010) Apoptosis and autophagy: decoding calcium signals that mediate life or death. Cold Spring Harb Perspect Biol 2:a005579
Kil J, Pierce C, Tran H, Gu R, Lynch ED (2007) Ebselen treatment reduces noise induced hearing loss via the mimicry and induction of glutathione peroxidase. Hear Res 226:44–51
Sha SH, Schacht J (2017) Emerging therapeutic interventions against noise-induced hearing loss. Expert Opin Investig Drugs 26:85–96
Sutherland GT, Chami B, Youssef P, Witting PK (2013) Oxidative stress in Alzheimer’s disease: primary villain or physiological by-product? Redox Rep 18:134–141
Kimberly CP, Mary H, Elizabeth RM, Beate RR (2019) Ambient air pollution, noise, and late-life cognitive decline and dementia risk. Annual Rev Public Health 40:203–220
Butterfield DA (2014) The 2013 SFRBM discovery award:selected discoveries from the butterfield laboratory of oxidative stress and its sequela in brain in cognitive disorders exemplified by Alzheimer disease and chemotherapy induced cognitive impairment. Free Rad Biol Med 74:157–174
Jafari Z, Kolb BE, Mohajerani MH (2020) Noise exposure accelerates the risk of cognitive impairment and Alzheimer’s disease: adulthood, gestational, and prenatal mechanistic evidence from animal studies. Neurosci Biobehav Rev 117:110–128. https://doi.org/10.1016/j.neubiorev.2019.04.001
Petrella C et al (2019) Neuropeptides in Alzheimer’s disease: an update. Curr Alzheimer Res 16:544–558
Matsui T et al (2007) Expression of APP pathway mRNAs and proteins in Alzheimer’s disease. Brain Res 1161:116–123
Guo T, Zhang D, Zeng Y et al (2020) Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol Neurodegeneration 15:40. https://doi.org/10.1186/s13024-020-00391-7
Tackenberg C, Nitsch RM (2019) The secreted APP ectodomain sAPPalpha, but not sAPPbeta, protects neurons against Abeta oligomer-induced dendritic spine loss and increased tau phosphorylation. Mol Brain 12:27
Zhang YW, Thompson R, Zhang H et al (2011) APP processing in Alzheimer’s disease. Mol Brain 4:3
Bibl M, Mollenhauer B, Lewczuk P, Esselmann H, Wolf S, Trenkwalder C, Otto M et al (2007) Validation of amyloid-beta peptides in CSF diagnosis of neurodegenerative dementias. Mol Psychiatry 12:671–680. https://doi.org/10.1038/sj.mp.4001967
Jan A, Gokce O, Luthi-Carter R, Lashuel HA (2008) The ratio of monomeric to aggregated forms of Abeta40 and Abeta42 is an important determinant of amyloid-beta aggregation, fibrillogenesis, and toxicity. J Biol Chem 17(283):28176–28189. https://doi.org/10.1074/jbc.M803159200
Willem M, Tahirovic S, Busche MA, Ovsepian SV, Chafai M et al (2015) (2015) Eta-secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 526:443–447
Gu Z, Liu W, Yan Z (2009) β-Amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. J Biol Chem 284:10639–10649
Bojarski L, Herms J, Kuznicki J (2008) Calcium dysregulation in Alzheimer’s disease. Neurochem Int 52:621–633
Van der Kant R, Goldstein LS, Ossenkoppele R (2020) Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci 21:21–35
Jafari Z, Mehla J, Kolb BE, Mohajerani MH (2019) Gestational stress augments postpartum β-amyloid pathology and cognitive decline in a mouse model of Alzheimer’s disease. Cereb Cortex 29:3712–3724. https://doi.org/10.1093/cercor/bhy251
Hradek AC, Lee HP, Siedlak SL, Torres SL, Jung W, Han AH, Lee HG (2015) Distinct chronology of neuronal cell cycle re-entry and tau pathology in the 3xTg-AD mouse model and Alzheimer’s disease patients. J Alzheimers 43:57–65
Nizzari M, Thellung S, Corsaro A et al (2012) Neurodegeneration in Alzheimer disease: role of amyloid precursor protein and presenilin 1 intracellular signaling. J Toxicol
Heneka MT, Carson MJ, El Khoury J et al (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14:388–405. https://doi.org/10.1016/S1474-4422(15)70016-5
Lue LF, Rydel R, Brigham EF, Yang LB, Hampel H et al (2001) Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia 35:72–79
Griffin WS, Nicoll JA, Grimaldi LM, Sheng JG, Mrak RE (2000) The pervasiveness of interleukin-1 in alzheimer pathogenesis: a role for specific polymorphisms in disease risk. Exp Gerontol 35:481–487. https://doi.org/10.1016/s0531-5565(00)00110-8
Wang WY, Tan MS, Yu JT, Tan L (2015) Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Translational Med 3:136
Solé M, Lenoir M, Durfort M et al (2021) Seagrass Posidonia is impaired by human-generated noise. Commun Biol 4:743. https://doi.org/10.1038/s42003-021-02165-3
Weuve J, D’Souza J, Beck T, Evans DA, Kaufman JD, Rajan KB, de Leon CFM, Adar SD (2021) Long-term community noise exposure in relation to dementia, cognition, and cognitive decline in older adults. Alzheimers Dement 17:525–533. https://doi.org/10.1002/alz.12191
Calderon-Garciduenas L, Solt AC, Henríquez-Roldán C, Torres-Jardón R, Nuse B et al (2008) Long-term air pollution exposure is associated with neuroinflammation, an altered innate immune response, disruption of the blood-brain barrier, ultrafine particulate deposition, and accumulation of amyloid beta-42 and alpha-synuclein in children and young adults. Toxicol Pathol 36:289–310
Kleinman MT, Araujo JA, Nel A, Sioutas C, Campbell A, Cong PQ, Li H, Bondy SC (2008) Inhaled ultrafine particulate matter affects CNS inflammatory processes and may act via MAP kinase signaling pathways. Toxicol Lett 178:127–130
Sirivelu MP, MohanKumar SM, Wagner JG, Harkema JR, MohanKumar PS (2006) Activation of the stress axis and neurochemical alterations in specific brain areas by concentrated ambient particle exposure with concomitant allergic airway disease. Environ Health Perspect 114:870–874
Daiber A, Kröller-Schön S, Oelze M, Hahad O, Li H, Schulz R, Steven S, Münzel T (2020) Oxidative stress and inflammation contribute to traffic noise-induced vascular and cerebral dysfunction via uncoupling of nitric oxide synthases. Redox Biol 34:101506
Cacciottolo M, Wang X, Driscoll I, Woodward N, Saffari A et al (2017) Particulate air pollutants, APOE alleles and their contributions to cognitive impairment in older women and to amyloidogenesis in experimental models. Transl Psychiatry 7:e1022
Lagouge M, Larsson NG (2013) The role of mitochondrial DNA mutations and free radicals in disease and ageing. J Intern Med 273:529–543
Dias V, Junn E, Mouradian MM (2013) The role of oxidative stress in Parkinson’s disease. J Parkinson’s Disease 3:461–491
Grimm MOW, Michaelson DM, Hartmann T (2017) Omega-3 fatty acids, lipids, and apoE lipidation in Alzheimer’s disease: a rationale for multi-nutrient dementia prevention. J Lipid Res 58:2083–2101
Duncan AL, Song W, Sansom MSP (2020) Lipid-dependent regulation of ion channels and G protein–coupled receptors: insights from structures and simulations. Annu Rev Pharmacol Toxicol 60:31–50
Munro S (2013) Lipid Rafts: Elusive or Illusive? Cell 115:377–388
Hicks D, Nalivaeva N, Turner A (2012) Lipid rafts and Alzheimer’s disease: protein-lipid interactions and perturbation of signaling. Front Physiol 3
Van der Kant R, Goldstein LSB (2015) Cellular functions of the amyloid precursor protein from development to dementia. Dev Cell 32:502–515
Kao Y-C, Ho P-C, Tu Y-K, Jou I-M, Tsai K-J (2020) Lipids and Alzheimer’s disease. Int J Mol Sci 21:1505. https://doi.org/10.3390/ijms21041505
Paul KC, Haan M, Mayeda ER, Ritz BR (2019) Ambient air pollution, noise, and late-life cognitive decline and dementia risk. Annu Rev Public Health 40:203–220
Ferreiro E, Oliveira CR, Pereira CM (2008) The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol Disease 30:331–342
Bonda DJ, Wang X, Perry G et al (2010) Oxidative stress in Alzheimer disease: a possibility for prevention. Neuropharmacology 59:290–294
Bonet-Costa V, Pomatto LC, Davies KJ (2016) The proteasome and oxidative stress in Alzheimer’s disease. Antioxid Redox Signal 25:886–901
Jack CR, Wiste HJ, Botha H, Weigand SD, Therneau TM, Knopman DS, Graff-Radford J, Jones DT, Ferman TJ, Boeve BF et al (2019) The bivariate distribution of amyloid-b and tau: relationship with established neurocognitive clinical syndromes. Brain 142:3230–3242
Pereira JB, Ossenkoppele R, Palmqvist S, Strandberg TO, Smith R, Westman E, Hansson O (2019) Amyloid and tau accumulate across distinct spatial networks and are differentially associated with brain connectivity. Elife 8:e50830
Griffiths TD, Lad M, Kumar S, Holmes E, McMurray B, Maguire EA, Billig AJ, Sedley W (2020) How can hearing loss cause dementia? Neuron 108:401–412. https://doi.org/10.1016/j.neuron.2020.08.003
Alexander N, Alexander DC, Barkhof F, Denaxas S (2021) Identifying and evaluating clinical subtypes of Alzheimer’s disease in care electronic health records using unsupervised machine learning. BMC Med Inform Decis Mak 21:343. https://doi.org/10.1186/s12911-021-01693-6
Ralli M, Gilardi A, Stadio AD, Severini C, Salzano FA, Greco A, Vincentiis M (2019) Hearing loss and Alzheimer’s disease: a review. Int Tinnitus J 23:79–85. https://doi.org/10.5935/0946-5448.20190014
Acknowledgements
We are grateful to Lilit Sukiasyan for assisting with writing and editing the material.
Author information
Authors and Affiliations
Contributions
Study concept and design, acquisition of data, analysis and interpretation of the data, drafting of the manuscript: Ashkhen Manukyan
Corresponding author
Ethics declarations
Ethical approval
None.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The author declares no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Manukyan, A.L. Noise as a cause of neurodegenerative disorders: molecular and cellular mechanisms. Neurol Sci 43, 2983–2993 (2022). https://doi.org/10.1007/s10072-022-05948-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-022-05948-6