
Abstract
Background
Amyotrophic lateral sclerosis (ALS) is a fatal and incurable neurodegenerative disease. There is still no established cost-effective treatment that can improve functional status and survival of ALS patients. Perampanel, by inhibiting neuronal calcium ion influx and preventing dyslocalization of nuclear proteins, has the potential to ameliorate ALS neurodegeneration.
Objectives
This study aims to determine the efficacy and safety of perampanel among ALS patients in terms of improvement in functional status using a review of relevant studies.
Methods
MedLine, Cochrane Central Register for Controlled Trials, Scopus, Embase, Literatura Latino-Americana e do Caribe em Ciências da Saúde, ClinicalTrials.gov website, and HERDIN databases were searched from inception to August 2021 for relevant studies.
Results
The search yielded 132 articles; 3 studies were included in the analysis. Pooled evidence shows that perampanel compared to placebo significantly improves cortical motor hyperexcitability but not the ALS functional rating scale-revised score. Perampanel is associated with adverse events such as aggression, somnolence, anger, and dysarthria.
Conclusion
There is no sufficient evidence to support the role of perampanel in improving functional status of ALS patients. Although it can ameliorate motor cortical hyperexcitability, its clinical benefit has not yet been elucidated. Perampanel is not well tolerated among ALS patients as it is associated with adverse events such as aggression, somnolence, anger, and dysarthria. Further studies investigating the role of perampanel early in the ALS disease course, excluding ALS patients with frontotemporal lobe degeneration features and C9ORF72 repeat expansion, and using gradual drug titration schedule are needed to evaluate the potential benefit of perampanel in ALS.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative syndrome characterized by progressive muscle weakness and atrophy [1, 2]. With a crude prevalence ranging from 1 to 11.3 per 100,000 population, ALS is the most common motor neuron disease [3, 4].
Several mechanisms have been implicated in the process of neurodegeneration in ALS—accumulation of ubiquitinated cytoplasmic inclusions in motor neurons [5], mutations in superoxide dismutase-1 (SOD-1) [6], and microglia-mediated neuro-inflammation [7]. Despite the ever-growing knowledge on the biologic processes involved in ALS and the continuous identification of potential pathogenic targets for drug development, promising therapy is still lacking [8]. As of this writing, only three drugs are known to improve survival or functional status of patients with ALS. Riluzole, a glutaminergic antagonist, is the first drug to be approved for the management of ALS in 1995 [9]. Edaravone, a free radical scavenger first trialed for cerebrovascular disease, is the most recent drug that has been given Food and Drug Administration approval for ALS [10]. Sodium phenylbutyrate-taurursodiol is an emerging drug for ALS [11]. Two recent trials demonstrated a significant beneficial effect of sodium phenylbutyrate-taurursodiol administration in terms of functional status [11] and survival [12] among patients with ALS. This drug is still yet to be approved for ALS.
Although recent trials have determined the efficacy of these drugs, issues on cost and logistics remain a fundamental concern. Several cost-effectiveness studies demonstrate inconsistent cost–utility profiles of prescribing these medications [13,14,15]. Hence, it is imperative to investigate an accessible and cost-effective drug with promising potential to improve functional status and survival of patients with ALS.
Perampanel, an approved drug for the treatment of partial and generalized seizures, has been the focus of recent studies on ALS. It is postulated that perampanel may prevent the cytoplasmic dyslocalization of TDP-43 protein by inhibiting calcium ion influx in motor neurons by non-competitive selective antagonism of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptors [16, 17]. This claim has been supported by a recent animal study which demonstrated that perampanel normalized TDP-43 pathology and prevented the emergence of the ALS phenotype in rodent models [17]. Recent clinical trials attempted to demonstrate the effect of perampanel on cortical motor thresholds as measured by transcranial magnetic stimulation [18], its safety and tolerability [19], and its effect on functional status [20] among patients with ALS.
This study aims to determine the efficacy and safety of perampanel among patients with ALS in terms of improvement in functional status using a review of relevant clinical studies.
Methods
The PRISMA (Preferred Reporting Items for Systematic reviews and Meta-analyses) consensus guidelines were followed in this review [21].
Criteria for selection of studies for this review
We considered randomized, double-blind, parallel group, placebo- and/or active-controlled clinical trials and quasi-experimental, cluster-randomized, cross-over, prospective or retrospective cohort, case–control, and cross-sectional studies in this review. We included studies involving patients who were diagnosed with ALS based on the El Escorial criteria and its revisions [22] or the Awaji criteria [23]. No restrictions in terms of age, sex, ethnicity, disease phenotype, and disease severity were employed. We included studies involving perampanel given per orem at any dose as the intervention. No restrictions in terms of concurrent or prior utilization of riluzole or edaravone were employed in the selection of studies. All studies tagged as primary research, reported in English, and available as full-text articles were included. Two reviewers (R1 and R2) screened all the titles, abstracts, and keywords independently to assess relevance to the objectives of this review. Discrepancies were reconciled by the third and fourth reviewers (R3 and R4) independently.
Outcome measures considered
-
Change in ALS Functional Rating Scale-Revised (ALSFRS-R) score—ALSFRS-R is a questionnaire-based test using a 0-to-48 scoring system with 1-point increments that measures functional status in terms of ability to perform activities of daily living [24]. A positive change means improvement in functional status.
-
Change in cortical excitability threshold—lowered cortical excitability threshold, a feature of ALS, can be assessed by measuring motor evoked potentials elicited by transcranial magnetic stimulation [18].
-
Adverse drug events (ADE)—the proportion of participants who experienced any serious and non-serious adverse drug event after drug administration assessed at a defined time.
Search methods for identification of studies and selection of studies
We searched the following databases for relevant studies: MEDLINE by PubMed, Cochrane Central Register for Controlled Trials (CENTRAL), Scopus, Embase, Literatura Latino-Americana e do Caribe em Ciências da Saúde (LILACS), ClinicalTrials.gov website, and HERDIN Database. The following general and MeSH term-based search strategy was employed: (perampanel OR 3-(2-cyanophenyl)-5-(2-pyridyl)-1-phenyl-1,2-dihydropyridin-2-one) AND (amyotrophic lateral sclerosis OR Gehrig’s Disease OR Gehrig Disease OR Gehrigs Disease OR Motor Neuron Disease OR Lou Gehrig’s Disease OR Lou-Gehrigs Disease OR Lou Gehrig Disease). Search strategies used in other databases are summarized in supplementary material.
Assessment of risk of bias, data collection, and analysis
The Cochrane Collaboration Tool was used in the assessment of risk bias of the included randomized trials. The following details were collected and collated appropriately from the included studies: study design, participants, intervention details for the treatment group and the control/placebo group, duration of treatment, and relevant outcomes described above.
Mean difference with 95% confidence intervals was used to measure treatment effect for the continuous outcomes, while risk difference (RD) of benefit or harm with 95% confidence intervals was used for the dichotomous outcomes. Syntheses of data were performed using the RevMan [Computer program] (version 5.4. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014).
Results
Included Studies
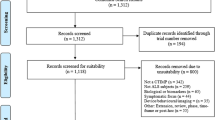
A total of 132 records were retrieved from major databases. Fifty-two records were identified as duplicates and were discarded. Eighty records were screened by two independent reviewers (R1 and R2), and 74 were excluded. Full-text copies of the remaining 6 articles were subjected to eligibility testing, and 3 records were excluded. A total of 3 articles were included in the qualitative and quantitative syntheses. Figure 1 shows the PRISMA flow diagram. Two studies were double-blind, randomized, placebo-controlled trials [18, 20], and the other study was an open-label pilot study [19]. Table 1 summarizes the characteristics of the included studies.

PRISMA flow diagram
Population characteristics in the included studies
A total of 90 patients were recruited in the studies. Table 2 summarizes the characteristics of patients in the included studies. Majority of the patients were elderly males with clinically probable or clinically probable laboratory-supported ALS who received both riluzole and edaravone as baseline therapies. The mean baseline ALSFRS-R score ranged from 28.5 to 40.3.
Interventions employed in the included studies
Different titration schedules were employed in the three included studies. In the trial by Aizawa and colleagues [20], respective full doses of perampanel were achieved via gradual dose escalation over 4 weeks. No strict titration schedule was implemented in the trial by Oskarsson and colleagues [18]—subjects assigned to the 4-mg perampanel group immediately received 4 mg per day dose, subjects assigned to the 8-mg perampanel group were returning patients who initially received 4 mg of perampanel per day for at least 3 weeks. In the open-label study by Hotait and colleagues [19], perampanel dose was increased by 2 mg per day per week until 8 mg per day was reached or until adverse effects were noted. The durations of treatment were 48 weeks, at least 3 weeks, and 12 weeks in the studies by Aizawa and colleagues, Oskarsson and colleagues, and Hotait and colleagues, respectively.
Assessment of risk of bias
Both trials are deemed to have low risk for selection, performance, and reporting biases as both employed randomized double-blind design. Both trials have unclear risk for attrition bias because of the considerable attrition rates of 18% and 50% in the placebo and treatment groups in the study by Aizawa and colleagues [20] and 25% in the treatment group in the study by Oskarsson and colleagues [18]. The study by Oskarsson is deemed to have unclear detection bias as it is not mentioned if the electrophysiologist who performed the transcranial magnetic stimulation measurements was blinded to the treatment allocation. Figure 2 shows the risk of bias summary.

Risk of bias summary of included trials
Effects of the intervention
Effect of perampanel on functional status of patients with ALS
In one trial evaluating the effect of perampanel on functional status using the ALSFRS-R, significant difference is observed in the mean difference of baseline and post-treatment ALSFRS-R scores between high-dose perampanel and placebo groups [mean difference = -8.4, 95% CI (-15.0 to -1.8), p = 0.0145] favoring the placebo group [20]. No significant difference is observed in the mean difference of baseline and post-treatment ALSFRS-R scores between low-dose perampanel and placebo groups in the same trial [mean difference = -4.5, 95% CI (-10.6 to 1.6), p = 0.1476] [20]. Upon dissecting the mean differences of the baseline and post-treatment ALSFRS-R sub-scores between high dose perampanel and placebo groups, only the bulbar sub-score shows a significant difference favoring placebo [mean difference = -2.7, 95% CI (-5.0 to -0.4), p = 0.0206]. Analysis of the mean differences in upper limbs, lower limbs, and respiratory sub-scores between the high-dose perampanel and placebo groups does not show significant differences with mean differences of -1.2 (95% CI -3.5 to 1.1, p = 0.3028), -1.2 (95% CI -3.4 to 0.9, p = 0.2583), and -1.4 (95% CI -3.2 to 0.5, p = 0.1375), respectively [20]. The open-label study by Hotait and colleagues [19] failed to demonstrate analysis of the effect of perampanel on ALSFRS-R scores as the study was prematurely terminated due to considerable emergence of adverse events.
Effectiveness of perampanel in increasing cortical excitability threshold among patients with ALS
The trial by Oskarsson and colleagues [18] demonstrates a statistically significant increase in cortical motor threshold from baseline to 2-h post-administration of perampanel by + 7.0% of maximal stimulator output (Q1, Q3; 3.8, 9.5; p = 0.02) in the low-dose (4 mg) group and by + 7.0% of maximal stimulator output (Q1, Q3; 4.0, 10.5; p < 0.01) in the combined low-dose and high-dose groups.
Safety of perampanel in patients with ALS
Pooled evidence from two clinical trials demonstrates that the proportion of patients who experienced any form of treatment-related adverse events is significantly higher in the perampanel group compared to the placebo group with risk difference of 0.48 (95% CI 0.33 to 0.63; p = 0.00001). Figure 3 summarizes pooled evidence on the rates of any adverse events in the perampanel and placebo groups. In the pilot study by Hotait [19], all six patients experienced adverse events—aggression (n = 5), somnolence (n = 3), anger (n = 3), dysarthria (n = 2), dizziness (n = 1), imbalance (n = 1), irritability (n = 1), and gait disturbance (n = 1) – which resolved upon discontinuation of the drug. In terms of serious adverse events, one trial reported the following events among patients randomized to the perampanel group (n = 43): dysphagia requiring gastrostomy (n = 10, 23%), gastric disturbance (n = 3, 7%), fracture / injury (n = 2, 5%), infection (n = 2, 5%), hallucination (n = 2, 5%), malignancy (n = 1, 2%), disseminated intravascular coagulation (n = 1, 2%), and death (n = 1, 2%).

Forest plot summarizing combinable data on treatment-related adverse drug effects from two trials
Discussion
This review provides comprehensive evidence from pooled findings of three studies on the efficacy and safety of perampanel in preventing cortical hyperexcitability and in delaying functional decline among patients with ALS.
The pathophysiologic mechanisms that suggested that perampanel may have a potential role in ALS are: (a) the cytoplasmic dyslocalization and accumulation of TDP-43, a protein involved in nuclear ribonucleic acid metabolism that is normally localized in the nuclear compartment [25], and (b) the abnormal increase in the density of calcium-permeable AMPA glutamate receptors on the membrane of motor neurons that promotes glutamate noise and increases the risk of excitotoxicity [26]. Both mechanisms lead to neurodegeneration and eventual cell death [27]. Perampanel, a non-competitive antagonist of AMPA glutamate receptor, is hypothesized to ameliorate neurodegeneration in ALS via its mechanism of reducing neuronal calcium ion influx, thereby reducing dyslocalization of TDP-43 protein in the cytoplasmic compartment and preventing eventual AMPA receptor-related excitotoxicity. This effect has been demonstrated in recent animal studies, which showed that the administration of perampanel normalized TDP-43 pathology, significantly prevented motor neuron death, and prevented the occurrence of ALS phenotype [17].
In humans, this potential role of perampanel on ALS was first demonstrated in the study by Oskarsson and colleagues [18]. They postulated that perampanel can reduce cortical hyperexcitability in ALS by reducing glutamate activation and neuronal calcium ion accumulation. Their findings showed that the administration of perampanel significantly increases the motor threshold—measured as the threshold at which 5 of 10 motor evoked potentials were elicited via transcranial magnetic stimulation—by 7% two hours after taking perampanel [18]. This finding, however, is reflective only of the acute electrophysiologic effect of the drug on motor neurons; its potential translation to clinical benefit is yet to be demonstrated. Two recent studies attempted to document the potential clinical benefit of perampanel in ALS in terms of functional status [18, 20].
The clinical trial by Aizawa and colleagues [20] failed to demonstrate clinical benefit of perampanel in terms of preventing functional status decline among patients with ALS. Rather, the study reveals that taking high-dose perampanel may significantly worsen bulbar sub-scores of patients with ALS. One possible reason is the non-concordance in pathophysiology. Although recent animal studies demonstrate that perampanel significantly ameliorates TDP-43 pathology in motor neurons [17], this does not take into consideration the other hypothesized patho-mechanisms involved in ALS, such as cytoplasmic accumulation of other proteins such as FUS [28], OPTN [29], C9ORF72 [30], UBQLN2) [31], and ATXN2 [32]; mitochondrial dysfunction leading to reactive oxygen species toxicity [6, 33]; and microglia-mediated release of pro-inflammatory cytokines [7]. It is the whole interplay of these genes, proteins, and pathways that directs the process of neurodegeneration and ultimately defines the clinical phenotype [34]. Another possible reason is the timing of administration. In the trial by Aizawa and colleagues [20] and the open-label study by Hotait and colleagues [19], the recruited patients already had an established ALS phenotype with mean illness duration of 13.11 months during the period of recruitment. Considering the proposed mechanism of perampanel in the prevention of neurodegeneration and eventual neuronal death, it can intuitively be proposed that this drug may have engendered clinical benefit if taken earlier in the course of the disease when upper and lower motor neuron dysfunctions are not yet florid. The open-label study by Hotait and colleagues [19] failed to demonstrate effect of perampanel on functional status of ALS patients, as majority of their recruited patients withdrew from the trial due to emergence of adverse events.
The tolerability and safety of perampanel among patients with ALS have been a cause of major concern in the recent trials. Recent studies on the safety profile of perampanel in patients with epilepsy show that the drug is fairly tolerated and is associated with relatively low incidence of serious adverse effects [35, 36]. Common adverse effects encountered by patients with epilepsy taking perampanel include dizziness, somnolence, aggressive behavior, and fatigue. Pooled evidence from this review demonstrates that the drug is not well tolerated among patients with ALS despite the adherence of the included studies to the recommended perampanel dosing. The most cited adverse event from pooled evidence is aggression. One possible reason for this occurrence is the inherent susceptibility of patients with ALS to behavioral changes due to its considerable overlap with frontotemporal lobar degeneration (FTLD) in terms of genetic basis, radiographic characteristics, and clinical features [37, 38]. In a population-based study done in Italy, 12.6% of ALS patients had overt FTLD and 37.2% had milder forms of impairment (executive cognitive impairment, non-executive cognitive impairment, behavioral impairment) or non-classifiable cognitive impairment [39]. FTLD, unlike Alzheimer disease and mixed dementias, is linked to high frequency of the apolipoprotein E e4 allele, a feature that is found to be associated with aggressive behavior [40]. This clinical association is also supported by findings of Grochmal-Bach and colleagues [41] in their study on aggression among patients with FTLD and Alzheimer disease. The evidence exploring the relationship between aggression and the milder forms of cognitive impairment in ALS is still lacking in the literature. It is also interesting to note that a particular subset of ALS patients presents a characteristically higher risk of developing FTLD. In an Irish population-based study, ALS patients carrying C9ORF72 hexanucleotide repeat expansion have significantly higher rate of FTLD (50%) than non-carriers (12%) [42]. Additionally, carriers were found to have a lower age of symptom onset, significant grey matter atrophy on imaging, strong ALF and FTLD family history, and ultimately, shorter survival. The prominence of cognitive and behavioral impairment among C9ORF72 repeat expansion carriers limits the potential of perampanel among these patients due to their inherent susceptibility to develop aggression.
The limitations of this review include the lack of study participants, with only 90 recruited patients from pooled studies. ALS is a heterogeneous condition with a wide range of clinical features that may not be fully represented in a small cohort. Another limitation of this review is the heterogeneity in terms of drug titration schedule and the duration of treatment. Certain adjustments may ameliorate these limitations.
The authors recommend that future studies should explore administering perampanel during the early stage of the disease, i.e., when corticomotor degeneration is not yet florid, to demonstrate the potential benefit of perampanel in preventing neuronal death. In terms of tolerability, the most implicated treatment-related adverse event is aggression. The authors deem this attributable to the inherent susceptibility of ALS patients to behavioral symptoms due to its significant overlap with frontotemporal lobe degeneration [37, 38], and perampanel might still be beneficial to ALS patients without the features of frontotemporal lobe degeneration. It is relevant to note that recent evidence suggests that even ALS patients who are classified to have normal cognition at initial presentation may develop significant cognitive decline and behavioral impairment later on. In an Italian longitudinal study involving 146 recruited ALS patients, 24% of cognitive normal patients on initial assessment developed significant cognitive decline and behavioral impairment after a median time of 7 months [43]. Nevertheless, none among those cognitive normal patients develop FTLD on follow-up assessment and the predominant behavioral complaint were depression and apathy on initial and follow-up evaluations, respectively [43]. Hence, the authors recommend that future studies should include detailed neuro-cognitive assessment and radiographic tests in screening ALS patients to exclude patients with features of frontotemporal lobar degeneration and those with C9ORF72 repeat expansion. The authors also suggest that future studies should implement a universal drug titration schedule to ameliorate treatment-related adverse events as these reactions were found to be dose-dependent [18, 20].
Conclusion
There is not enough evidence to support the role of perampanel at a dose of 4 or 8 mg per day among patients with ALS in improving functional status. Although pooled evidence suggests that perampanel can improve motor cortical hyperexcitability as measured by motor evoked potentials elicited by transcranial magnetic stimulation, its translation to clinical benefit has not yet been elucidated. Perampanel administration seems to be not very well tolerated among patients with ALS, as it is associated with adverse events such as aggression, somnolence, anger, and dysarthria.
Further studies investigating the potential role of perampanel early in the disease course of ALS, excluding ALS patients with frontotemporal lobe degeneration features and those with C9ORF72 repeat expansion, and using gradual drug titration schedule are needed to evaluate the potential benefit of perampanel in ALS.
References
Hardiman O, Al-Chalabi A, Chio A et al (2017) Amyotrophic lateral sclerosis. Nat Rev Dis Prim 3:17071. https://doi.org/10.1038/nrdp.2017.71
van Es MA, Hardiman O, Chio A et al (2017) Amyotrophic lateral sclerosis. Lancet 390:2084–2098. https://doi.org/10.1016/S0140-6736(17)31287-4
Chiò A, Logroscino G, Traynor BJ et al (2013) Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 41:118–130. https://doi.org/10.1159/000351153
Foster LA, Salajegheh MK (2019) Motor neuron disease: pathophysiology, diagnosis, and management. Am J Med 132:32–37. https://doi.org/10.1016/j.amjmed.2018.07.012
Blokhuis AM, Groen EJN, Koppers M et al (2013) Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol 125:777–794. https://doi.org/10.1007/s00401-013-1125-6
Gordon P (2013) Amyotrophic lateral sclerosis: an update for 2013 clinical features, pathophysiology, management and therapeutic trials. Aging Dis 04:295–310. https://doi.org/10.14336/AD.2013.0400295
Heneka MT, Kummer MP, Latz E (2014) Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14:463–477. https://doi.org/10.1038/nri3705
Kiernan MC, Vucic S, Talbot K et al (2021) Improving clinical trial outcomes in amyotrophic lateral sclerosis. Nat Rev Neurol 17:104–118. https://doi.org/10.1038/s41582-020-00434-z
Miller RG, Mitchell JD, Moore DH (2012) Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD001447.pub3
Abe K, Aoki M, Tsuji S et al (2017) Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 16:505–512. https://doi.org/10.1016/S1474-4422(17)30115-1
Paganoni S, Macklin EA, Hendrix S et al (2020) Trial of sodium Phenylbutyrate-Taurursodiol for amyotrophic lateral sclerosis. N Engl J Med 383:919–930. https://doi.org/10.1056/NEJMoa1916945
Paganoni S, Hendrix S, Dickson SP et al (2021) Long-term survival of participants in the <scp>CENTAUR</scp> trial of sodium phenylbutyrate-taurursodiol in <scp>amyotrophic lateral sclerosis</scp>. Muscle Nerve 63:31–39. https://doi.org/10.1002/mus.27091
Tavakoli M, Malek M (2001) The cost utility analysis of riluzole for the treatment of amyotrophic lateral sclerosis in the UK. J Neurol Sci 191:95–102. https://doi.org/10.1016/S0022-510X(01)00618-9
Messori A, Trippoli S, Becagli P, Zaccara G (1999) Cost effectiveness of Riluzole in amyotrophic lateral sclerosis. Pharmacoeconomics 16:153–163. https://doi.org/10.2165/00019053-199916020-00004
Yeo CJJ, Simmons Z (2018) Discussing edaravone with the ALS patient: an ethical framework from a U.S. perspective. Amyotroph Lateral Scler Front Degener 19:167–172. https://doi.org/10.1080/21678421.2018.1425455
Sills GJ, Rogawski MA (2020) Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 168:107966. https://doi.org/10.1016/j.neuropharm.2020.107966
Akamatsu M, Yamashita T, Hirose N et al (2016) The AMPA receptor antagonist perampanel robustly rescues amyotrophic lateral sclerosis (ALS) pathology in sporadic ALS model mice. Sci Rep 6:28649. https://doi.org/10.1038/srep28649
Oskarsson B, Mauricio EA, Shah JS et al (2021) Cortical excitability threshold can be increased by the AMPA blocker Perampanel in amyotrophic lateral sclerosis. Muscle Nerve 64:215–219. https://doi.org/10.1002/mus.27328
Hotait M, Ismail HH, Saab GE, Salameh JS (2021) An open label pilot study of the safety and tolerability of Perampanel in amyotrophic lateral sclerosis. Muscle Nerve 64(4):504–508. https://doi.org/10.1002/mus.27385
Aizawa H, Kato H, Oba K et al (2021) Randomized phase 2 study of perampanel for sporadic amyotrophic lateral sclerosis. J Neurol. https://doi.org/10.1007/s00415-021-10670-y
Moher D, Liberati A, Tetzlaff J, Altman DG (2009) Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med 6:e1000097. https://doi.org/10.1371/journal.pmed.1000097
Ludolph A, Drory V, Hardiman O et al (2015) A revision of the El Escorial criteria - 2015. Amyotroph Lateral Scler Front Degener 16:291–292. https://doi.org/10.3109/21678421.2015.1049183
Nodera H, Izumi Y, Kaji R (2007) New diagnostic criteria of ALS (Awaji criteria). Brain Nerve 59:1023–1029. https://doi.org/10.11477/mf.1416100142
Cedarbaum JM, Stambler N, Malta E et al (1999) The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. J Neurol Sci 169:13–21. https://doi.org/10.1016/S0022-510X(99)00210-5
Mackenzie IRA, Bigio EH, Ince PG et al (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61:427–434. https://doi.org/10.1002/ana.21147
Guo C, Ma Y-Y (2021) Calcium Permeable-AMPA receptors and excitotoxicity in neurological disorders. Front Neural Circuits 15:711564. https://doi.org/10.3389/fncir.2021.711564
Barmada SJ, Skibinski G, Korb E et al (2010) Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci 30:639–649. https://doi.org/10.1523/JNEUROSCI.4988-09.2010
Lagier-Tourenne C, Polymenidou M, Cleveland DW (2010) TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 19:R46–R64. https://doi.org/10.1093/hmg/ddq137
Osawa T, Mizuno Y, Fujita Y et al (2011) Optineurin in neurodegenerative diseases. Neuropathology 31:569–574. https://doi.org/10.1111/j.1440-1789.2011.01199.x
Renton AE, Majounie E, Waite A et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257–268. https://doi.org/10.1016/j.neuron.2011.09.010
Deng H-X, Chen W, Hong S-T et al (2011) Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477:211–215. https://doi.org/10.1038/nature10353
Elden AC, Kim H-J, Hart MP et al (2010) Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466:1069–1075. https://doi.org/10.1038/nature09320
Jaiswal MK (2017) Riluzole but not melatonin ameliorates acute motor neuron degeneration and moderately inhibits SOD1-mediated excitotoxicity induced disrupted mitochondrial Ca2+ signaling in amyotrophic lateral sclerosis. Front Cell Neurosci 10:295. https://doi.org/10.3389/fncel.2016.00295
Keon M, Musrie B, Dinger M et al (2021) Destination amyotrophic lateral sclerosis. Front Neurol 12:596006. https://doi.org/10.3389/fneur.2021.596006
Youn SE, Kim SH, Ko A et al (2018) Adverse events during perampanel adjunctive therapy in intractable epilepsy. J Clin Neurol 14:296. https://doi.org/10.3988/jcn.2018.14.3.296
Rugg-Gunn F (2014) Adverse effects and safety profile of perampanel: a review of pooled data. Epilepsia 55:13–15. https://doi.org/10.1111/epi.12504
Abramzon YA, Fratta P, Traynor BJ, Chia R (2020) The overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front Neurosci 14:42. https://doi.org/10.3389/fnins.2020.00042
Ling S-C, Polymenidou M, Cleveland DW (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79:416–438. https://doi.org/10.1016/j.neuron.2013.07.033
Montuschi A, Iazzolino B, Calvo A et al (2015) Cognitive correlates in amyotrophic lateral sclerosis: a population-based study in Italy. J Neurol Neurosurg Psychiatry 86:168–173. https://doi.org/10.1136/jnnp-2013-307223
Gotovac K, NikolacPerković M, Pivac N, Borovečki F (2016) Biomarkers of aggression in dementia. Prog Neuro-Psychopharmacology Biol Psychiatry 69:125–130. https://doi.org/10.1016/j.pnpbp.2016.03.002
Grochmal-Bach B, Bidzan L, Pachalska M et al (2009) Aggressive and impulsive behaviors in Frontotemporal dementia and Alzheimer’s disease. Med Sci Monit 15:CR248-54
Byrne S, Elamin M, Bede P et al (2012) Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol 11:232–240. https://doi.org/10.1016/S1474-4422(12)70014-5
Bersano E, Sarnelli MF, Solara V et al (2020) Decline of cognitive and behavioral functions in amyotrophic lateral sclerosis: a longitudinal study. Amyotroph Lateral Scler Front Degener 21:373–379. https://doi.org/10.1080/21678421.2020.1771732
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None. There are no conflicts of interest to declare in this research.
Ethical approval and informed consent
No ethical approval is needed because data from published studies in which informed consent was obtained by investigators were retrieved and analysed.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Turalde, C.W.R., Moalong, K.M.C., Espiritu, A.I. et al. Perampanel for amyotrophic lateral sclerosis: A systematic review and meta-analysis. Neurol Sci 43, 889–897 (2022). https://doi.org/10.1007/s10072-022-05867-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-022-05867-6