
Abstract
The autonomic nervous system (ANS), hypothalamic–pituitary–adrenal (HPA) axis, and immune system are connected anatomically and functionally. These three systems coordinate the central and peripheral response to perceived and systemic stress signals. Both the parasympathetic and sympathetic components of the autonomic nervous system rapidly respond to stress signals, while the hypothalamic–pituitary–adrenal axis and immune system have delayed but prolonged actions. In vitro, animal, and human studies have demonstrated consistent anti-inflammatory effects of parasympathetic activity. In contrast, sympathetic activity exerts context-dependent effects on immune signaling and has been associated with both increased and decreased inflammation. The location of sympathetic action, adrenergic receptor subtype, and timing of activity in relation to disease progression all influence the ultimate impact on immune signaling. This article reviews the brain circuitry, peripheral connections, and chemical messengers that enable communication between the ANS, HPA axis, and immune system. We describe findings of in vitro and animal studies that challenge the immune system with lipopolysaccharide. Next, neuroimmune connections in animal models of chronic inflammatory disease are reviewed. Finally, we discuss how a greater understanding of the ANS-HPA-immune network may lead to the development of novel therapeutic strategies that are focused on modulation of the sympathetic and parasympathetic nervous system.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The autonomic nervous system (ANS) plays a crucial role in maintaining homeostasis through its connections to multiple physiologic systems including the hypothalamic-pituitary-adrenocortical (HPA) axis and the immune system. Although the relationships between the ANS and some systems, such as the cardiovascular system, are understood in significant detail, there is still much to be understood about how the ANS, HPA axis, and the immune system coordinate a response to stress. Through both the parasympathetic nervous system (PNS) and sympathetic nervous system (SNS), the ANS can reflexively respond to a stress signal within seconds. In contrast, the HPA axis and immune system take longer to respond and typically have more prolonged actions.
This review addresses the brain circuitry, peripheral connections, and chemical messengers underlying an integrated ANS-HPA-immune network. The first section reviews the structural circuitry of the ANS, and its connections to HPA axis, and immune system. The second section focuses on the chemical messengers and receptors that underlie the functional connectivity of the network. The final section reviews clinical studies that demonstrate a wide array of diseases that are associated with a dysregulated ANS-HPA-immune network (Fig. 1).

Bidirectional anatomical and functional connections exist between the autonomic nervous system (ANS), hypothalamic–pituitary–adrenal (HPA) axis, and immune system
Anatomy
In this section, important components of the ANS and its structural connections with the HPA axis and immune system will be reviewed. Our understanding of the ANS-HPA-immune circuitry has been advanced by transneuronal labeling [1, 2], a technique that involves a microinjection of neurotropic live viruses into peripheral tissue or brain of the study animal. Budding viruses are transported to continuous neurons across synapses. Immunohistochemistry (IHC) techniques for viral products enable visualization of primary, secondary, or third order neurons [1, 2].
Structures of the autonomic nervous system (ANS)
Structures subserving autonomic function span the entire neuroaxis, from the brain to peripheral nerves, and beyond to diverse target organs. The term central autonomic network (CAN) typically refers to cortical and subcortical brain regions that regulate autonomic function [3]. Important structures include the nucleus of the solitary tract (NTS), insula cortex, hypothalamus, amygdala, prefrontal cortex, hippocampus, and periaqueductal gray (PAG) [1, 4]. In the brainstem, the NTS is the first relay station for visceral afferent projections and sends information through multi-synaptic pathways to the hypothalamus as well as limbic structures including the hippocampus and prefrontal cortex [5]. The hypothalamus is a brain regional critical to the stress response and maintenance of homeostasis. It addition to information from the NTS, the hypothalamus receives signals regarding the internal and external environmental from forebrain structures, including the primary viscero-motor and sensory cortex located in the insular cortex. Limbic structures including the prefrontal cortex, amygdala, and hippocampus link emotions to the ANS. The hippocampus receives multi-synaptic input from brainstem nuclei and has connections to the paraventricular nucleus (PVN) of the hypothalamus [5]. Finally, the PAG receive connections from the aforementioned forebrain structures, the brainstem, and the spinal cord and coordinate autonomic and somatic responses to stress and pain [5].
In contrast to the highly integrated connections of the CAN, caudally there is a structural and functional divergence into parasympathetic, sympathetic, afferent, and enteric components of the ANS [6]. The SNS is complex and physiologically diverse, sending projections to all organ systems in the body. Preganglionic SNS neurons reside in the lateral horns of the thoracolumbar segments and project to nearby prevertebral, paravertebral, and previsceral ganglia. The post-ganglionic fibers of the SNS innervate smooth muscle, cardiac muscle, and parenchymal organs, including all organs of the immune system. The parasympathetic division originates from nuclei in the brainstem and in the sacral spinal cord [6].
The vagus nerve, the main component of the parasympathetic nervous system, is a bidirectional nerve bundle that originates in the medulla and contains both efferent and afferent fibers [7, 8]. Important vagal efferents innervate the cardiovascular and gastrointestinal system to modulate heart rate and gastrointestinal motility. Vagal afferents comprise approximately 80% of the vagus nerve and carry visceral information from almost every organ system to the NTS [9].
The enteric nervous system (ENS) resides within the walls of the gut and modulates gastrointestinal function [10]. The ENS receives important cholinergic projections from gastrointestinal branches of the vagus nerve. Vagal efferents stimulate gastric acid production, promote motility, and modulate immune cell activity within gastrointestinal mucosa [10]. Gut vagal afferent fibers play an important role in the “gut-brain axis” and influence the brain circuitry involved in mood and anxiety disorders [9].
Anatomic connections between the ANS and the immune system
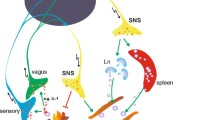
The ANS has structural connections to the organs of the immune system which are categorized as primary and secondary lymphoid organs. The thymus and bone marrow are primary lymphoid organs and produce lymphocytes. Secondary lymphoid organs include the lymph nodes, spleen, and tonsils, as well as mucosal tissue of the skin, gut, lungs, and peritoneum [11]. Both primary and secondary lymphoid organs receive significant sympathetic innervation (please see [12] for detailed review). In brief, transneuronal labeling studies have shown sympathetic innervation to the thymus originates in sympathetic premotor neurons in the hypothalamus, pons, and medulla oblongata. These premotor neurons project to sympathetic preganglionic neurons in the intermediolateral (IML) cell column of the T1-T7 spinal cord. Pre-ganglionic neurons in the IML synapse at nearby sympathetic ganglia. Post-ganglionic SNS projections innervate the thymus [12]. Similarly, in the case of bone marrow, premotor sympathetic brain nuclei in the hypothalamus, pons, and brain stem project to T8-L1 spinal sympathetic preganglionic neurons, with post-ganglionic projections innervating the bone marrow. Immunohistochemistry (IHC) studies have found no evidence of vagal or sensory innervation to the thymus or bone marrow [13,14,15].
Sympathetic innervation has also been studied in secondary lymphoid organs, including the spleen and lymph nodes. In animal models, neuronal mapping has demonstrated that the spleen receives input from prevertebral sympathetic ganglia associated with the celiac-mesenteric plexus and the thoracic sympathetic chain through the splenic nerve [13]. While there is no evidence for parasympathetic or sensory input to the spleen in numerous rodent models [13,14,15], a retrograde mapping study of the cat splenic nerve using horseradish peroxidase demonstrated labeling in the dorsal root ganglion, indicating the cat splenic nerve contains sensory afferents [16]. In lymph nodes, SNS innervation originates from the postganglionic neurons that supply sympathetic input to the area of the body where the lymph node resides (e.g., the superior cervical lymph node receives innervation from the ipsilateral superior cervical ganglion) [17]. Recently, advanced techniques using optogenetics, RNA sequencing, and retrograde labeling have demonstrated that lymph nodes receive sensory innervation, and this innervation has a preference for the LN periphery, where inflammation-related cellular changes are most likely to occur [18].
Anatomic connections between the ANS and the HPA axis
The HPA axis and the ANS are anatomically connected through the hypothalamus (Fig. 2). On the efferent side, the hypothalamus can trigger the rapid sympatho-adrenomedullary (SAM) pathway, as well as the slower HPA axis. On the afferent side, the NTS receives vagal projections that detect systemic stress signals (e.g., interleukins, hypoglycemia, pain signals). These data are conveyed, via adrenergic and noradrenergic projections, to the PVN of the hypothalamus, directly activating the HPA axis [19]. Destruction of these ascending NTS neurons reduces the HPA axis response to systemic perturbations such as an interleukin-1 beta (IL-1 β) injection and hypoglycemia [20], but does not impact the HPA axis response to perceived emotional stressors. Therefore, ascending catecholaminergic pathways appear to specifically relay afferent detected systemic stress signals and are not necessary for perceived stress to influence HPA axis output. Communication between the ANS and HPA axis is bidirectional. The PVN activates the SNS and PNS through projections to the IML and dorsal motor nucleus of the vagus (DMV), respectively [19]. The presence of both parasympathetic and sympathetic projection neurons in the PVN supports its role as a central coordinator of the ANS [21].

Stress signals, detected by afferents, and perceived stress, detected by the forebrain, are processed through bottom-up and top-down circuits that activate the hypothalamic pituitary adrenal (HPA)-axis and autonomic nervous system (ANS)
Activity of the HPA axis and ANS is also influenced by top-down processing that encodes perceived or emotional stress. This emotional-autonomic network has been identified through both animal tracing and human imaging studies and includes the hippocampus, central nucleus of the amygdala, and bed nucleus of the stria terminalis. The hippocampus has been identified as a region of particular importance in activating both the ANS and HPA [3]. A direct projection from the ventral subiculum of the hippocampus to the PVN has been identified through retrograde transport studies. Activation of glucocorticoid receptors (GR) expressed in the hippocampus result in decreased activity of the HPA axis [22]. The hippocampus also receives information from the main vagal relay station, the NTS, through a multi-synaptic pathway, indicating that the hippocampus receives information regarding both perceived and afferent-detected stressors [5].
Physiology
In this section, we will describe the chemical messengers that enable communication between the ANS, HPA axis, and immune system.
Neurohumoral interactions between the ANS and the immune system
Both the SNS and PNS have chemical connections to the immune system. In the PNS, both pre- and post-ganglionic parasympathetic fibers use acetylcholine (ACh) as their main neurotransmitter. ACh binds to muscarinic (mAChR) and nicotinic receptors (nAChr) that are each comprised of several subunits that heterodimerize to enable cell and tissue specificity [23]. Immune cells have both muscarinic and nicotinic receptors. A receptor subtype, which appears to be particularly important for immune signaling modulation, is the α7ChR. This receptor exists on innate immune cells including macrophages, dendritic cells, and T and B cells and plays an essential role in the suppression of cytokine synthesis through the cholinergic anti-inflammatory pathway (discussed further in “Animal models” section).
In the SNS, pre-synaptic fibers use acetylcholine as their neurotransmitter, while post-ganglionic sympathetic fibers use norepinephrine (NE), with few exceptions [24]. NE binding of β and α adrenergic receptors (AR) on immune cells triggers second messenger systems including protein-kinase-A (PKA) and the mitogen-activated protein kinase (MAPK) pathways. Activation of these second messenger pathways modulate cytokine release and alter immune cell differentiation, leading to pro- or anti- inflammatory activity [25]. Activation of α-AR leads to an increase in pro-inflammatory cytokines tumor necrosis factor alpha (TNF-α) and IL-6, while the activation of β-AR is associated with lower levels of inflammation [26]. Anti-inflammatory action occurs through inhibition of pro-inflammatory cytokines or promotion of anti-inflammatory cytokines such as IL-1ra, TNF-receptor fragments, and IL-10 [26]. Relative α/β-AR expression and differences in NE binding affinity for α/β-ARs permit context-dependent effects of SNS activity on immune cell signaling [25].
In addition to post-ganglionic release of NE directly on lymphoid organs and immune cells, NE is released into the blood stream following activation of the sympatho-adrenomedullary (SAM) axis by the hypothalamus following a stress [27]. Systemically, NE has indirect actions on the immune system through regulation of blood and lymph flow, recruitment of inflammatory cells, and stimulation of leukocyte generation in the bone marrow [28]. The concentration of NE in the systemic blood stream is lower than the concentration of NE found at the junction of post-ganglionic fibers and α/β-AR. NE has a higher binding affinity at α-AR. Thus, it has been posited that systemically circulating NE acts predominantly at α-AR, while NE released by post-ganglionic projections acts at both α-AR and β-AR [25]. Therefore, the location of NE activity contributes to the context-dependent influences of the SNS on the immune system.
Neurohumoral interactions between the ANS and the HPA axis
The HPA axis and ANS interact at several levels of the neuroendocrine axis through their effector molecules, i.e., glucocorticoids in the case of the HPA axis and norepinephrine for the ANS. The majority of studies demonstrate that catecholamines (including norepinephrine) activate the HPA axis. Dual labeling IHC studies have demonstrated that catecholamine-synthesizing afferents, originating in the NTS, terminate on cells in the PVN that release corticotropin releasing factor (CRF). CRF travels in the hypophyseal portal system to stimulate the anterior pituitary gland to release adrenocorticotropic hormone (ACTH), which in turn stimulates the release of glucocorticoids from the adrenal glands [29]. Activation of the SAM pathway also results in systemic release of catecholamines that act on ARs in the PVN to stimulate the HPA axis [30].
The mechanisms by which the HPA axis influences ANS activity are less well understood but likely involve reduced parasympathetic activity and cardiovagal baroreflex sensitivity. In animals, administration of glucocorticoids in the NTS rapidly decreased the activity of baroreceptive neurons and depressed cardiovagal baroreflex sensitivity [31]. This result was reproduced in human subjects [32]. A bolus of hydrocortisone acutely increased heart rate and blood pressure and reduced cardiovagal baroreflex sensitivity and heart rate variability in healthy young males [32]. While this mechanism is still under investigation, glucocorticoid modulation of serotonergic signaling between the prefrontal cortex and the amygdala through 5HT1A receptors may play an important role [33].
Pathophysiology
In this section, we describe how the ANS, HPA axis, and immune system communicate during acute and chronic stress. First, we will first describe findings of in vitro and animal studies that use lipopolysaccharide (LPS) to acutely challenge the immune system. LPS is an endotoxin present on gram-negative bacteria which triggers the innate immune response. Next, neuroimmune connections in animal models of more chronic inflammatory disease will be reviewed. Finally, human observational and interventional studies examining the therapeutic potential of PNS and SNS modulation will be discussed.
In vitro studies
To understand the role of the PNS in inflammation, α7nAChR has been studied in immune cells exposed to LPS. Both macrophages and dendritic cells treated with ACh release less TNF-α, IL-6, and other pro-inflammatory cytokines in response to LPS through actions of ACh at α7-nAChR [34, 35]. Through α7-nAChR, ACh also suppresses macrophage production of LPS-induced matrix metalloproteinases (MMP)-9, an activator of cytokines and chemokines. [36]
Following an LPS challenge, the SNS can have pro- or anti-inflammatory effects on immune signaling that depend on AR subtype [37]. As seen in the resting state (described in Sect. 3.1), α-AR activity is pro-inflammatory, while β-AR is anti-inflammatory [12, 38]. Stimulation of macrophage α-ARs with an α-agonist or a low concentration of NE (that preferentially activates α-AR) increased the production of LPS-induced TNF-α [39]. This effect was prevented by the α-AR antagonist yohimbine [39]. In contrast, NE action at macrophage β-ARs decreased LPS-induced TNF-α. The augmentation of TNF-α by NE was inhibited by a β-blocker [12, 38].
Animal models
Experimental models of endotoxemia have measured the effect of physical manipulation of the vagus nerve (i.e., vagal nerve stimulation or vagotomy), genetic manipulation of cholinergic receptors, or pharmacologic stimuli (cholinergic or anticholinergic) on the response to an inflammatory stimulus. Vagal afferents play a critical role in the detection and transmission of systemic stress signals to the brain. Rodents post vagotomy did not display the central increase in IL-1β expression following a peripheral injection of LPS [40, 41]. The brain may also initiate a response to stress signals through vagal efferents. Following LPS administration, stimulation of the peripheral vagus nerve resulted in attenuated serum TNF-α production and a lower likelihood of septic shock [35]. The anti-inflammatory effect of vagal nerve stimulation (VNS) has been demonstrated in other models of acute inflammatory stress including experimental pancreatitis [42] and myocardial ischemia/reperfusion injury [43]. In models of chronic disease including inflammatory bowel disease [44], arthritis [45], and cardiovascular disease [46], VNS reduces inflammation and disease severity. The precise anti-inflammatory mechanism is incompletely understood and is not purely parasympathetic, as sympathetic intermediaries are required [47]. While diminished TNF-α release from splenic macrophages is a broadly accepted part of the “cholinergic anti-inflammatory pathway” (CAP), there is no evidence for vagal innervation of the spleen. The necessity of splenic adrenergic neurons, ACh-producing T cells, and the α7ChR has been established in animal models, as vagal stimulation did not produce TNF-α suppression in mice without functional T cells, mice lacking α7ChR, and mice post splenectomy [48]. One working model proposes that vagal efferents may influence adrenergic mesenteric ganglia supplying post-ganglionic projections to the spleen through the splenic nerve [49]. Sympathetic innervation may activate β2AR on splenic T cells to release ACh and activate α7ChR on splenic macrophages which results in decreased production of TNF-α [34]. A rodent model of immune-mediated arthritis provides another example of how vagal activity reduces inflammation through a circuitry involving the SNS and stimulation of β2AR [50, 51].
The impact of systemic and local SNS action on immune signaling and disease progression has been studied in animal models of inflammatory disorders. NE released into the bloodstream primarily acts through pro-inflammatory α-AR [52]. Local release of NE by post-ganglionic fibers on target tissue (e.g., synovial fluid, hepatocytes, adipose tissue, or intestine) can act at β-AR or α-AR. In a rodent model of immune-mediated hepatitis, activation of β2AR produces anti-inflammatory effects [53, 54]. Receptor expression levels, sensitivity, and concentration of NE determine the balance of α /β-AR activity, and thus, pro-/anti-inflammatory impact of adrenergic modulation. Disease severity at time of SNS modulation is another important contextual factor that can determine the ultimate consequence of SNS activity on immune signaling [55].
Human studies
The ANS-HPA-immune network has been studied in humans through observational and interventional studies. Observational studies have typically examined ANS activity in patients with chronic inflammatory disorders or have made associations between immune signaling molecules and ANS activity. Interventional studies have used both vagus nerve stimulation and pharmacological agents to modulate ANS signaling in patients with inflammatory diseases.
Observational studies in humans
Basic tenets of cardiac physiology have driven the design of many observational studies. Heart rate variability (HRV), defined as the fluctuation in length of sequential R-R intervals, is determined by parasympathetic and sympathetic efferent activity to the heart [56]. Reduced HRV is a marker of dysautonomia and is associated with many chronic diseases [56]. The balance of sympathetic and parasympathetic input can be assessed through time and frequency-domain analyses of HRV. The high-frequency (HF) component of HRV is associated with vagally mediated activity and flexibility of the ANS, while the low-frequency (LF) component of HRV has been theorized to reflect more general ANS activity, but with a sympathetic predominance [57]. The majority of studies examining the relationship between the ANS and immune signaling have utilized indices of resting HRV. A more limited body of work has used measures of reflexive HRV including changes in heart rate in response to stimuli such as valsalva maneuver (VM), deep breathing, and tilt-table testing.
In observational studies, parasympathetically mediated indices of cardiac function are negatively associated with both markers of inflammation and chronic inflammatory diseases, while those associated with SNS activity are positively associated with markers of inflammation and chronic inflammatory diseases [58]. In a recent meta-analysis, the parasympathetically mediated HF-HRV and standard deviation of R-R intervals (SDNN) had the strongest correlations (negative direction of effect) with markers of inflammation [58]. The association between low vagal activity and vulnerability to inflammation is present early in life. In pre-term infants, for every one standard deviation decrease in HF-HRV, the odds ratio of developing necrotizing enterocolitis (NEC) increased ten-fold [59]. In adults, patients with inflammatory bowel disease (IBD) had lower HF-HRV compared to healthy controls. In adults, rheumatoid arthritis (RA), IBD, and neuropsychiatric conditions are all associated with cardiac measurements indicative of lower resting vagal activity [58]. Using measures of reflexive HRV, our lab has demonstrated that vagal dysfunction in patients living with HIV is associated with small intestinal bacterial overgrowth (SIBO), which is linked to elevation of the pro-inflammatory cytokine IL-6 [60].
Studies focused on the SNS in humans are methodologically more complex for reasons related to the physiology and pathophysiology of the system itself and issues of measurement. The SNS is a tonically active system and as such its dysfunction may involve under- or over-activity or a combination thereof. For example, a hyperadrenergic state has been described in early diabetic autonomic neuropathy prior to development of more overt autonomic failure [61]. Moreover, physical and psychological stressors and the activity of the HPA axis influence short-term SNS activity. Methodologic challenges center upon the difficulty of directly measuring SNS activity. Microneurography, although technically challenging, can directly record muscle sympathetic nerve activity (MSNA) but is limited by significant floor effects. Sympathetic nerve fiber density provides detailed local information but requires collection of tissue specimen. Thus, many studies use heart rate and blood pressure-based parameters, including LF-HRV (as described above), and adrenergic baroreflex sensitivity (BRSA), a continuous variable defined as the rate of systolic blood pressure recovery (mmHg/second) following release of the VM. Drawbacks include dependence on patient effort during the VM and confounding effects of medications.
Congruent with the signal transduction pathways identified through in vitro and in vivo models, human observational studies have shown context-dependent influences of adrenergic signaling during inflammation. An acute, robust increase in NE concentration acts through β-AR to reduce inflammation. In acute sepsis, NE infusion has an anti-inflammatory effect that correlates with the infusion rate and can be inhibited with a β-blocker [62]. Similarly, a hyperventilation technique that significantly increased resting NE and LPS-induced NE resulted in reduced production of IL-6, IL-8, and TNF-α in healthy controls [63]. In contrast, when the inflammation state persists, as occurs in many chronic inflammatory illnesses, systemic SNS activity is positively correlated with inflammation. [64] For example, increased SNS activity in hypertension is associated with cardiac inflammation and remodeling [65]. In patients with RA, general sympathetic activity as measured by MSNA was positively correlated with C-reactive protein (CRP) [64]. The pro-inflammatory influence of NE in states of chronic inflammation may be related to downregulation and desensitization of immune cell β2-ARs following chronic stimulation that favors pro-inflammatory α-AR signaling [52]. In chronic diseases, there is also local repulsion of SNS fibers from immune organs, reducing NE concentration at target tissue and the local anti-inflammatory action at β-AR [64].
Human interventional studies
There has been significant interest in treating inflammatory conditions through modulation of vagal nerve activity [66]. In a small pilot study on patients with mild and moderate Crohn’s disease study, surgically implanted VNS, or “invasive” VNS (iVNS), resulted in symptomatic improvement in the majority of patients [67]. Further, following incubation with LPS, blood samples from Crohn’s patients who received iVNS had a reduction in TNF-α levels [68]. A non-invasive handheld VNS device (nVNS, gammaCore) was FDA-cleared for the treatment of two inflammatory disorders, migraine and cluster headache disorders [69]. In women with Sjogren’s syndrome, nVNS for 4 weeks was associated with reductions in IL-1β and TNF-α levels [70]. In patients with refractory gastroparesis, almost half experienced symptomatic improvement with the use of nVNS 2–3 times daily [71]. In our own lab, we found that 8 weeks of the acetylcholinesterase inhibitor, pyridostigmine, resulted in improved small intestinal bacterial overgrowth (SIBO) and decreased levels of pro-inflammatory cytokines for our patients with HIV-autonomic neuropathy [72].
The majority of therapeutic interventions targeting the ANS have focused on increasing cholinergic signaling. However, studies that have targeted the SNS have also shown promising results. In a small randomized double blind study, sympathetic ganglionectomy decreased pain for patients with RA [73]. The analgesic effect of β blockers has been examined for the treatment of surgical pain. An infusion of esmolol during surgery was found to decrease post-operative pain severity and opioid consumption [74, 75]. Patients with active UC, given 8 weeks of transdermal clonidine, had a reduction in sympathetic activity as measured by MSNA, plasma epinephrine levels, and spectral indices of cardiac sympathetic activity. The reduction of sympathetic overactivity correlated to reduction in UC disease activity [76].
Remaining questions and future directions
The anatomically and functionally interconnected ANS-HPA-immune plays an important, but incompletely understood role in inflammatory disease development. In this final section, we pose questions that reflect current gaps in our understanding of the ANS-HPA-Immune network.
-
1)
What role does the SNS play in the transition from a state of acute to chronic inflammation and can modulation of the SNS early in disease prevent inflammatory chronification? While the majority of therapeutic advances have focused on modulating the PNS, modulating the SNS during key windows of disease vulnerability may prevent a state of chronic inflammation. The animal models comparing the efficacy of sympathectomy in early and late stages of RA suggest that the timing of SNS modulation is critical. By preventing chronic inflammation, SNS modulation may also be a promising analgesic. Viewing pain as a state of dysregulated ANS-HPA-immune signaling sheds light on the putative analgesic mechanisms of angiotensin receptor blockade and alpha/beta blockade in migraine treatment [77].
-
2)
How do sex hormones influence the ANS-HPA-immune network? Few studies have examined sex differences in the ANS-HPA-immune network. There are sex differences in the autonomic nervous system [78] and HPA axis [68]. Estrogen has been shown to decrease sympathetic nerve discharge by ~ 30%. Further, injection of estrogen into areas of the CAN including the NTS and medulla also decreases sympathetic activity and heart rate, suggesting both peripheral and central actions of estrogen on the ANS [79]. This is an opportunity for future studies to advance our understanding of the female predominance of chronic inflammatory diseases.
-
3)
Does the hippocampus provide the anatomical substrate for the link between disorders of central inflammation, dysautonomia, and a dysregulated HPA axis? The hippocampus’s modulation of the ANS and HPA axis, as well as its plasticity, make it an important, yet understudied, coordination center of the HPA axis, ANS, and immune systems through mechanisms that are not yet well understood [16]. Hippocampal atrophy is present in a diverse array of disorders associated with low-grade inflammation and dysautonomia. However, few studies have examined the role of the hippocampus in coordinating the ANS-HPA-immune system [80].
By addressing these questions, we hope future studies will lead to the development of novel therapeutic strategies for our patients suffering from diseases that involve a dysregulated ANS-HPA-immune network.
Availability of data and material
Not applicable.
Code availability
Not applicable.
References
Loewy AD (1998) Viruses as transneuronal tracers for defining neural circuits. Neurosci Biobehav Rev 22(6):679–684
Ugolini G (2020) Viruses in connectomics: viral transneuronal tracers and genetically modified recombinants as neuroscience research tools. J Neurosci Methods 346:108917
Harrison NA et al (2013) Central autonomic network mediates cardiovascular responses to acute inflammation: relevance to increased cardiovascular risk in depression? Brain Behav Immun 31:189–196
Thome J et al (2017) Desynchronization of autonomic response and central autonomic network connectivity in posttraumatic stress disorder. Hum Brain Mapp 38(1):27–40
Castle M, Comoli E, Loewy AD (2005) Autonomic brainstem nuclei are linked to the hippocampus. Neuroscience 134(2):657–669
Ondicova K, Mravec B (2010) Multilevel interactions between the sympathetic and parasympathetic nervous systems: a minireview. Endocr Regul 44(2):69–75
Shields RW Jr (1993) Functional anatomy of the autonomic nervous system. J Clin Neurophysiol 10(1):2–13
Pembroke P (1971) The anatomy of the vagus nerve in the cervical area in man. J Anat 110(Pt 3):502
Berthoud HR, Neuhuber WL (2000) Functional and chemical anatomy of the afferent vagal system. Auton Neurosci 85(1–3):1–17
Spencer NJ, Hu H (2020) Enteric nervous system: sensory transduction, neural circuits and gastrointestinal motility. Nat Rev Gastroenterol Hepatol 17(6):338–351
Abbas AK, Lichtman AH, Pillai S (2019) Basic immunology: functions and disorders of the immune system, 4th edn. Elsevier, Philadelphia
Nance DM, Sanders VM (2007) Autonomic innervation and regulation of the immune system (1987–2007). Brain Behav Immun 21(6):736–745
Nance DM, Burns J (1989) Innervation of the spleen in the rat: evidence for absence of afferent innervation. Brain Behav Immun 3(4):281–290
Bellinger DL et al (1993) Acetylcholinesterase staining and choline acetyltransferase activity in the young adult rat spleen: lack of evidence for cholinergic innervation. Brain Behav Immun 7(3):191–204
Schafer MK, Eiden LE, Weihe E (1998) Cholinergic neurons and terminal fields revealed by immunohistochemistry for the vesicular acetylcholine transporter. II. The peripheral nervous system. Neuroscience 84(2):361–376
Baron R, Janig W (1988) Sympathetic and afferent neurons projecting in the splenic nerve of the cat. Neurosci Lett 94(1–2):109–113
Panuncio AL et al (1999) Adrenergic innervation in reactive human lymph nodes. J Anat 194(Pt 1):143–146
Huang S et al (2021) Lymph nodes are innervated by a unique population of sensory neurons with immunomodulatory potential. Cell 184(2):441-459 e25
Swanson LW, Kuypers HG (1980) The paraventricular nucleus of the hypothalamus: cytoarchitectonic subdivisions and organization of projections to the pituitary, dorsal vagal complex, and spinal cord as demonstrated by retrograde fluorescence double-labeling methods. J. Comp Neurol 194:555–570
Buller K et al (2001) Dorsal and ventral medullary catecholamine cell groups contribute differentially to systemic interleukin-1beta-induced hypothalamic pituitary adrenal axis responses. Neuroendocrinology 73(2):129–138
Kreier F et al (2006) Tracing from fat tissue, liver, and pancreas: a neuroanatomical framework for the role of the brain in type 2 diabetes. Endocrinology 147(3):1140–1147
Westerhaus MJ, Loewy AD (2001) Central representation of the sympathetic nervous system in the cerebral cortex. Brain Res 903(1–2):117–127
Bering B, Moises HW, Muller WE (1987) Muscarinic cholinergic receptors on intact human lymphocytes. Properties and subclass characterization. Biol Psychiatry 22(12):1451–1458
Benarroch EE (1994) Neuropeptides in the sympathetic system: presence, plasticity, modulation, and implications. Ann Neurol 36(1):6–13
Pongratz G, Straub RH (2014) The sympathetic nervous response in inflammation. Arthritis Res Ther 16(6):504
Schneemilch CE, Bank U (2001) Release of pro- and anti-inflammatory cytokines during different anesthesia procedures. Anaesthesiol Reanim 26(1):4–10
Elenkov IJ et al (2000) The sympathetic nerve–an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev 52(4):595–638
Powell ND et al (2013) Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via beta-adrenergic induction of myelopoiesis. Proc Natl Acad Sci U S A 110(41):16574–16579
Valenta LJ, Elias AN, Eisenberg H (1986) ACTH stimulation of adrenal epinephrine and norepinephrine release. Horm Res 23(1):16–20
Toufexis DJ, Walker CD (1996) Noradrenergic facilitation of the adrenocorticotropin response to stress is absent during lactation in the rat. Brain Res 737(1–2):71–77
Ouyang M, Wang S (2000) Dexamethasone attenuates the depressor response induced by neuropeptide Y microinjected into the nucleus tractus solitarius in rats. Br J Pharmacol 129(5):865–870
Adlan AM et al (2018) Acute hydrocortisone administration reduces cardiovagal baroreflex sensitivity and heart rate variability in young men. J Physiol 596(20):4847–4861
Agorastos A et al (2019) Vagal effects of endocrine HPA axis challenges on resting autonomic activity assessed by heart rate variability measures in healthy humans. Psychoneuroendocrinology 102:196–203
Wang H et al (2003) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421(6921):384–388
Borovikova LV et al (2000) Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405(6785):458–462
Yang YH et al (2015) Acetylcholine inhibits LPS-induced MMP-9 production and cell migration via the alpha7 nAChR-JAK2/STAT3 pathway in RAW264.7 cells. Cell Physiol Biochem 36(5):2025–38
Li W et al (2003) Regulation of noradrenergic function by inflammatory cytokines and depolarization. J Neurochem 86(3):774–783
Kin NW, Sanders VM (2006) It takes nerve to tell T and B cells what to do. J Leukoc Biol 79(6):1093–1104
Spengler RN et al (1990) Stimulation of alpha-adrenergic receptor augments the production of macrophage-derived tumor necrosis factor. J Immunol 145(5):1430–1434
Maier SF et al (1998) The role of the vagus nerve in cytokine-to-brain communication. Ann N Y Acad Sci 840:289–300
Li S et al (2018) Intestinal microbiota impact sepsis associated encephalopathy via the vagus nerve. Neurosci Lett 662:98–104
van Westerloo DJ et al (2006) The vagus nerve and nicotinic receptors modulate experimental pancreatitis severity in mice. Gastroenterology 130(6):1822–1830
Mioni C et al (2005) Activation of an efferent cholinergic pathway produces strong protection against myocardial ischemia/reperfusion injury in rats. Crit Care Med 33(11):2621–2628
Jin H et al (2017) Anti-inflammatory effects and mechanisms of vagal nerve stimulation combined with electroacupuncture in a rodent model of TNBS-induced colitis. Am J Physiol Gastrointest Liver Physiol 313(3):G192–G202
Koopman FA et al (2017) Balancing the autonomic nervous system to reduce inflammation in rheumatoid arthritis. J Intern Med 282(1):64–75
Chapleau MW et al (2016) Chronic vagal nerve stimulation prevents high-salt diet-induced endothelial dysfunction and aortic stiffening in stroke-prone spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 311(1):H276–H285
Martelli D et al (2014) Reflex control of inflammation by sympathetic nerves, not the vagus. J Physiol 592(7):1677–1686
Matteoli G et al (2014) A distinct vagal anti-inflammatory pathway modulates intestinal muscularis resident macrophages independent of the spleen. Gut 63(6):938–948
Martelli D, McKinley MJ, McAllen RM (2014) The cholinergic anti-inflammatory pathway: a critical review. Auton Neurosci 182:65–69
Janig W, Green PG (2014) Acute inflammation in the joint: its control by the sympathetic nervous system and by neuroendocrine systems. Auton Neurosci 182:42–54
Miao FJ, Levine JD (1999) Neural and endocrine mechanisms mediating noxious stimulus-induced inhibition of bradykinin plasma extravasation in the rat. J Pharmacol Exp Ther 291(3):1028–1037
Bellinger DL, Lorton D (2018) Sympathetic nerve hyperactivity in the spleen: causal for nonpathogenic-driven chronic immune-mediated inflammatory diseases (IMIDs)? Int J Mol Sci 19(4):1188. https://doi.org/10.3390/ijms19041188
Tiegs G, Bang R, Neuhuber WL (1999) Requirement of peptidergic sensory innervation for disease activity in murine models of immune hepatitis and protection by beta-adrenergic stimulation. J Neuroimmunol 96(2):131–143
Neuhuber WL, Tiegs G (2004) Innervation of immune cells: evidence for neuroimmunomodulation in the liver. Anat Rec A Discov Mol Cell Evol Biol 280(1):884–892
Harle P et al (2005) An opposing time-dependent immune-modulating effect of the sympathetic nervous system conferred by altering the cytokine profile in the local lymph nodes and spleen of mice with type II collagen-induced arthritis. Arthritis Rheum 52(4):1305–1313
Sosnowski M et al (2005) Heart rate variability fraction–a new reportable measure of 24-hour R-R interval variation. Ann Noninvasive Electrocardiol 10(1):7–15
Rajendra Acharya U et al (2006) Heart rate variability: a review. Med Biol Eng Comput 44(12):1031–1051
Williams DP et al (2019) Heart rate variability and inflammation: a meta-analysis of human studies. Brain Behav Immun 80:219–226
Doheny KK et al (2014) Diminished vagal tone is a predictive biomarker of necrotizing enterocolitis-risk in preterm infants. Neurogastroenterol Motil 26(6):832–840
Robinson-Papp J et al (2018) Vagal dysfunction and small intestinal bacterial overgrowth: novel pathways to chronic inflammation in HIV. AIDS 32(9):1147–1156
Jacob G, Costa F, Biaggioni I (2003) Spectrum of autonomic cardiovascular neuropathy in diabetes. Diabetes Care 26(7):2174–2180
Stolk RF et al (2020) Norepinephrine dysregulates the immune response and compromises host defense during sepsis. Am J Respir Crit Care Med 202(6):830–842
Kox M et al (2014) Voluntary activation of the sympathetic nervous system and attenuation of the innate immune response in humans. Proc Natl Acad Sci U S A 111(20):7379–7384
Miller LE et al (2000) The loss of sympathetic nerve fibers in the synovial tissue of patients with rheumatoid arthritis is accompanied by increased norepinephrine release from synovial macrophages. FASEB J 14(13):2097–2107
Levick SP et al (2010) Sympathetic nervous system modulation of inflammation and remodeling in the hypertensive heart. Hypertension 55(2):270–276
Bonaz B, Sinniger V, Pellissier S (2016) Anti-inflammatory properties of the vagus nerve: potential therapeutic implications of vagus nerve stimulation. J Physiol 594(20):5781–5790
Pellissier S et al (2010) Psychological adjustment and autonomic disturbances in inflammatory bowel diseases and irritable bowel syndrome. Psychoneuroendocrinology 35(5):653–662
Goel N et al (2014) Sex differences in the HPA axis. Compr Physiol 4(3):1121–1155
Yuan H, Silberstein SD (2016) Vagus nerve and vagus nerve stimulation, a comprehensive review: Part I. Headache 56(1):71–78
Tarn J et al (2019) The effects of noninvasive vagus nerve stimulation on fatigue and immune responses in patients with primary Sjögren’s syndrome. Neuromodulation 22(5):580–585
Paulon E et al (2017) Proof of concept: short-term non-invasive cervical vagus nerve stimulation in patients with drug-refractory gastroparesis. Frontline Gastroenterol 8(4):325–330
Robinson-Papp J et al (2019) The effect of pyridostigmine on small intestinal bacterial overgrowth (SIBO) and plasma inflammatory biomarkers in HIV-associated autonomic neuropathies. J Neurovirol 25(4):551–559
Kidd BL et al (1992) Role of the sympathetic nervous system in chronic joint pain and inflammation. Ann Rheum Dis 51(11):1188–1191
Vahabi S, Rafieian Y, Abbas Zadeh A (2018) The effects of intraoperative esmolol infusion on the postoperative pain and hemodynamic stability after rhinoplasty. J Invest Surg 31(2):82–88
Mendonca FT et al (2021) Intra-operative esmolol and pain following mastectomy: A randomised clinical trial. Eur J Anaesthesiol 38(7):735–743
Furlan R et al (2006) Sympathetic overactivity in active ulcerative colitis: effects of clonidine. Am J Physiol Regul Integr Comp Physiol 290(1):R224–R232
Halker RB et al (2016) ACE and ARB Agents in the prophylactic therapy of migraine-how effective are they? Curr Treat Options Neurol 18(4):15
Koenig J, Thayer JF (2016) Sex differences in healthy human heart rate variability: a meta-analysis. Neurosci Biobehav Rev 64:288–310
Mercuro G et al (2000) Evidence of a role of endogenous estrogen in the modulation of autonomic nervous system. Am J Cardiol 85(6):787–9, A9
Vinkers CH et al (2021) An integrated approach to understand biological stress system dysregulation across depressive and anxiety disorders. J Affect Disord 283:139–146
Acknowledgements
We thank Celestine He for her contribution to this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
All authors provide their consent to publish.
Conflict of Interest/Competing interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mueller, B., Figueroa, A. & Robinson-Papp, J. Structural and functional connections between the autonomic nervous system, hypothalamic–pituitary–adrenal axis, and the immune system: a context and time dependent stress response network. Neurol Sci 43, 951–960 (2022). https://doi.org/10.1007/s10072-021-05810-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-021-05810-1