
Abstract
This is the second part of a two-part document intended to discuss recent therapeutic progresses in genetic neuromuscular disorders. The present review is for diseases of motor neuron and skeletal muscle, some of which reached recently the most innovative therapeutic approaches. Nusinersen, an SMN2 mRNA splicing modifier, was approved as first-ever therapy of spinal muscular atrophy (SMA) by FDA in 2016 and by EMA in 2017. The orally administered small-molecule risdiplam, which increases SMN protein levels similarly but also in peripheral organs, is tested in ongoing phase 2 and 3 trials. After positive results with phase 1 treatment with AAV9-SMN, the first gene therapy for SMA, a phase 3 clinical trial is ongoing. Ataluren is the first approved drug for Duchenne muscular dystrophy (DMD) patients with premature stop codon mutations and its indication has been recently extended since the age of 2 years. Exon skipping technology was and is currently tested in many phase 3 trials, and eteplirsen received a conditional approval by FDA for patients amenable to exon 51 skipping, but not by EMA. Many other compounds with different mechanisms of action are now tested in DMD by phase 2 and 3 trials, including phase 1 gene therapy. Other innovative approaches are under investigation, i.e., gene therapy in X-linked myotubular myopathy and Pompe disease, and antisense oligonucleotides in myotonic dystrophy type 1. Positive evidences are discussed about lamotrigine and ranolazine in non-dystrophic myotonias, chaperons in Pompe disease, and nucleosides in mitochondrial DNA depletion induced by thymidine kinase 2 deficiency.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
This is the second part of a two-part document intended to review recent therapeutic progresses in genetic neuromuscular disorders. Part 1 is dedicated to peripheral neuropathies, whereas the present review is for diseases of motor neuron and skeletal muscle, which reached recently the most advanced development of new, innovative, often mutation-specific, therapeutic approaches. Methods used include splicing modification by antisense oligonucleotides (ASOs), readthrough of premature stop codons, use of viral vectors to introduce correct genetic information, or enhancing the effectiveness of enzyme replacement therapies [1].
Inherited motor neuron and muscle disorders are complex diseases with special requirements and need of a multiprofessional care. Improvement of cardiac and/or respiratory function has produced an increasing life expectancy in diseases such as spinal muscular atrophy (SMA) and Duchenne muscular dystrophy (DMD). On the other hand, there are now new medical complications being reported which had not previously been seen, and transition from pediatric to adult age enhances even more the demand of innovative care models [2,3,4]. Here, we review the most clinically significant therapeutic progresses which are going to modify completely the natural history of these disorders.
Spinal muscular atrophy
SMA, linked to chromosome 5q and with autosomal recessive inheritance, includes disorders which are different in age of onset and severity. They all are characterized by degeneration of alpha motor neurons in the spinal cord with progressive muscle atrophy and weakness. SMA type 1 (SMA1) is the most severe form with onset in the first 6 months of age, no acquisition of the sitting position, and usually death in the first years. SMA type 2 (SMA2) patients have disease onset between 6 and 18 months and acquire sitting position but do not walk unless with support. SMA type 3 (SMA3) and type 4 (SMA4) are the mildest forms, with onset after 18 months and in the second-third decade, respectively, independent walking but variable clinical course. SMA patients require a multidisciplinary assistance which has been recently reviewed with an update on diagnosis, pulmonary management, rehabilitation, orthopedic and spinal management, nutritional, swallowing and gastrointestinal care, acute care, and ethical issues [4, 5].
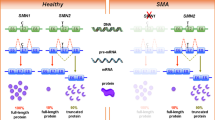
The peculiar genetic alteration of SMA is characterized by loss or mutation of both copies of the survival motor neuron 1 (SMN1) gene, which is not compensated by presence of nearly identical paralog SMN2. SMN2 gene harbors a silent mutation in exon 7 that alters the splicing of the mRNA leading to the predominant production of a truncated, unstable protein along with a minority of correctly spliced transcripts, generating low levels (around 10%) of full-length protein. Moreover, SMN2 copy number varies in individuals, being the most important phenotypic modifier of the disease. Determination of SMN2 copy number is essential to establish careful genotype–phenotype correlations, predict disease evolution, and stratify patients for clinical trials, higher SMN2 copy number being associated with prediction of more benign evolution [6].
Nusinersen
Without any doubt, SMA is the disease with the most innovative and effective therapies discovered in the last few years [7]. Although olesoxime, a neuroprotective and neuroregenerative agent, had shown some clinical benefits in maintaining motor function in SMA2 and SMA3 patients over a 24-month period in a double-blind, placebo-controlled phase 2 study [8], the therapeutic scenario has been subverted by nusinersen. It is an ASO that binds to the SMN2 pre-mRNA downstream of exon 7, leading to the translation of a fully functional SMN protein. After evidence of increased SMN protein expression in treated mice spinal cord motor neurons, phase 1, 2, and 3 trials have been performed with intrathecal drug administration since it does not cross blood-brain barrier. In both randomized, double-blind, sham-controlled, phase 3 trials in infants with onset at 2–20 weeks (infantile-onset SMA; ENDEAR trial) and at 6–20 months (later-onset SMA; CHERISH trial), respectively, interim analysis results prompted early termination, because of ethical considerations for the infants in the control group. In the former, treated patients showed improvements in motor function and were alive without the use of permanent assisted ventilation compared with those undergoing a sham procedure, and some of them achieved meaningful motor milestones [9]. Significant improvement in motor function was also achieved in later-onset SMA after nusinersen treatment [10]. The incidence of all adverse events was similar in the nusinersen group and the control group in both trials, as well as that of moderate or severe adverse events in CHERISH, or even higher in the controls in ENDEAR trial. Nobody discontinued treatment because of an adverse event in CHERISH, and adverse events leading to discontinuation of nusinersen/sham procedure were even lower in nusinersen group in ENDEAR. Lumbar puncture (LP) related events such as procedural and post-LP back pain, headache, and post-LP syndrome occurred below 15% in later-onset SMA trial [9, 10].
In the United States of America (USA), the Food and Drug Administration (FDA) approved the drug in December 2016 and the European Medicines Agency (EMA) in June 2017. Nusinersen became available firstly with an expanded access program in select territories for approximately 1 year before becoming available on the market with the commercial name of Spinraza® (Biogen Inc.) [11]. A phase 2 clinical trial with nusinersen is still ongoing in 25 infants who were genetically diagnosed with presymptomatic SMA and also began treatment in the presymptomatic stage of the disease (ClinicalTrials.gov, NCT02386553). In October 2018, interim analysis showed that all patients were alive and none required tracheostomy or permanent ventilation (https://spinalnewsinternational.com/nurture-spinraza-sma/). Additionally, 17 participants were able to walk independently, and all 25 were able to sit without support. This study confirms that early diagnosis and treatment with nusinersen has the potential to change the course of SMA. Positive changes have been even recorded in some SMA1 patients outside the age range of the inclusion criteria of the original trial, i.e., starting nusinersen when they were older than 2 years and even older than 10 years [12]. In July 2018, Biogen informed about the occurrence of communicating hydrocephalus not related to meningitis or bleeding in rare patients treated with Spinraza®, which needs urgent medical attention to any possible early symptoms or signs [13].
Risdiplam
Risdiplam (RG7916, RO7034067) is an orally administered, centrally and peripherally distributed, small-molecule SMN2 mRNA splicing modifier, developed by F. Hoffmann-La Roche, PTC Therapeutics and SMA Foundation for the treatment of SMA. It promotes similar increases of SMN protein levels in the brain and peripheral organs of SMA mice [14]. After demonstration of safety and tolerability in a phase 1 study in male volunteers with escalating dose [15], still ongoing phase 2 and 3 clinical trials started in SMA1, 2, and 3 patients (NCT02913482, NCT02908685, NCT03032172). In December 2018, a Roche media release announced that EMA has granted PRIME (PRIority Medicines) designation for risdiplam. PRIME designation is granted to support data generation and development plans for promising medicines, providing a pathway for accelerated evaluation by EMA and thus potentially reaching patients earlier. In the same release, Roche announced that trials’ interim analysis showed that treated SMA1 infants met developmental milestones, including sitting without support. Nineteen of 21 patients remained alive. No infant required tracheostomy or permanent ventilation and none has lost the ability to swallow. Interim data from SMA2 and 3 trial demonstrated a median greater than twofold increase in SMN protein levels in the blood following 12 months of treatment and a median 3.1-point improvement in Motor Function Measure in 30 patients treated with risdiplam for at least 1 year. To date, there have been no drug-related safety findings leading to withdrawal from the studies (https://www.roche.com/media/releases/med-cor-2018-12-17.htm). An open-label, single-arm, multicenter clinical study is also going to start to investigate the efficacy, safety, pharmacokinetics, and pharmacodinamics of risdiplam in babies with genetically diagnosed and presymptomatic SMA (NCT03779334).
SMN1 gene replacement
A long duration pre-clinical research has led to demonstration that a single intravenous injection of adeno-associated virus-9 (AAV9) delivering SMN gene (AAV9-SMN) to replace the missing/mutated SMN1 gene is sufficient to rescue the disease phenotype in SMA mouse model. Moreover, 10 times lower dose of AAV9-SMN delivered directly to the cerebral spinal fluid via single injection produces widespread transgene expression throughout the spinal cord in mice and nonhuman primates [16]. Phase 1 treatment with AAV9-SMN, named AVXS-101 (Zolgensma®, Avexis Inc.), in the first ever gene therapy trial in SMA1 babies, was associated with an increased survival rate compared to the normal course of the disease and the achievement and maintenance of motor milestones that SMA1 infants normally would not be expected to achieve [17]. The most commonly observed side effect of AVXS-101 was elevated liver enzymes. Treated patients have also reduced pulmonary and nutritional support requirements and decreased hospitalization rate over a 2-year follow-up period [18]. A phase 3, open-label, single-arm, single-dose, gene replacement therapy clinical trial with AVXS-101 in SMA1 is ongoing in USA and Europe (NCT03306277, NCT03461289). In December 2018, Novartis, the parent company of Avexis Inc., announced that FDA has accepted the company’s Biologics License Application (BLA) under Priority Review for Zolgensma®. Priority Review status requires the FDA to review the application and decide on whether to approve the drug within 6 months. Moreover, a phase 1 trial of AVXS-101 in SMA type 2 is now fully enrolled (December 2018) and results are expected by May 2019 (NCT03381729).
Duchenne muscular dystrophy
DMD is a severe progressive X-linked disorder due to the lack of the protein dystrophin. Deletions, duplications, and point mutations in the dystrophin gene stop the transduction of dystrophin protein. Therefore, the link between cytoskeleton and extracellular matrix is lost, leading to instability of muscle membrane and cell necrosis followed by an exhaustible regeneration. Cell necrosis parallels to a robust inflammatory response involving different factors such as MAPK, COX, LOX, leukotriene B-4, TNF-α, reactive oxygen species, and nuclear factor-κB and Hippo signaling pathways, which are considered possible therapeutic targets [19,20,21,22]. This process produces the increase of adipose and connective tissue leading to progressive muscle weakness. Cognitive and motor developmental milestones can be acquired with delay, and this is a common presentation of the disorder. The average age at diagnosis is still around 4–5 years of age and can vary among countries, with a significant delay between symptoms onset and genetic diagnosis [23].
Since diagnosis but especially after patients become wheelchair-bound by a median age of 12 years, a multidisciplinary approach is mandatory with regard to the respiratory, cardiological, orthopedic, psychosocial, and nutritional needs [3, 24, 25]. Moreover, transition from pediatric to adult life is a fragile period for the management of complications as most adult medical specialists are not used to care DMD patients [26], as well as for different personal needs such as independent social life. The use of inspiratory and expiratory supports decreases the number of hospitalizations, and the use of nocturnal ventilation significantly prolongs life expectancy in ventilated patients [27]. Despite this, cardiomyopathy remains a major cause of morbidity and mortality. The lack of symptoms due to limited motor function often causes a late referral to a cardiac specialist contributing to a poor clinical outcome. Joint contractures, scoliosis, need of spinal surgery, osteoporosis, and risk of vertebral fractures need a careful management. Finally, nutritional complications include obesity, malnutrition, swallowing difficulties, and constipation [28].
Steroids are now part of care recommendations for DMD, since they improve muscle strength and function in the short term of 12 months and strength up to 2 years [28]. However, adverse effects include weight gain, behavioral abnormalities, cushingoid appearance, excessive hair growth, osteoporosis, cataract, and increased risk of bone fractures [29]. The best corticosteroid regime has still to be defined, and an international study, FOR-DMD (NCT01603407), is ongoing and aims to compare the three most frequently prescribed regimens of corticosteroids: prednisolone 0.75 mg/kg/day 10 days on and 10 days off, prednisolone 0.75 mg/kg/day, and deflazacort 0.9 mg/kg/day [30].
Ataluren
Ataluren (Translarna™, PTC Therap.), the first approved drug for DMD, is an oral drug which can be prescribed in patients with premature stop codon mutations. Point mutations introducing a premature stop codon into mRNA and therefore causing the translation of a truncated and non-functional protein, are present in 10–15% of DMD patients [31]. Ataluren binds ribosomal RNA subunits and impedes the recognition of premature stop codons. In August 2014, EMA gave to ataluren a conditional approval for the treatment of DMD ambulatory patients aged 5 years and older with nonsense mutations. ACT DMD, a multicenter, randomized, double-blind, placebo-controlled, phase 3 trial (NCT01826487), had tested the safety and efficacy of ataluren, administered orally three times daily, in ambulatory boys aged 7–16 years with nonsense mutation DMD. The primary endpoint, distance change at 6-min walk test (6MWT), was in favor of ataluren in the intent-to-treat population analysis, although it did not reach the statistical significance (mean change − 47.7 m in treated vs − 60.7 m in placebo patients). In the pre-specified subgroup with baseline 6MWT values between 300 and 400 m, the difference was significant in favor of ataluren-treated patients vs placebo (− 27.0 m vs − 69.9 m; p = 0.007, respectively) [32]. After 48 weeks of treatment, the number of non-ambulatory patients was lower in the treated group compared to controls (8% vs 12%) and in the pre-specified ≥ 300 to < 400 m subgroup (none vs 8%). Ataluren was generally well tolerated, and most treatment-emergent adverse events were mild to moderate in severity.
A clinical trial assessing the long-term outcomes of ataluren in patients with nonsense mutation DMD is currently ongoing (NCT03179631). It is a randomized, double-blind, placebo-controlled, 72-week study followed by a 72-week open-label period. The completion date for this study is December 2021. In October 2018, PTC Therap. announced preliminary data from the first international drug registry for DMD patients receiving Translarna™, highlighting the long-term clinical benefit in delaying irreversible muscle loss when compared with published natural history. In the real-world setting, DMD children and adolescents are continuing to walk years longer than untreated children and are staying more physically able. A time-to-event analysis for loss of ambulation in treated subjects has shown a median age of loss of ambulation of 16.5 years of age (https://www.multivu.com/players/English/8420051-ptc-therapeutics-stride-registry-duchenne-muscular-dystrophy-translarna/). Based on the positive results of a phase 2 study in DMD children from 2 to 5 years, in May 2018, EMA has extended the indication of ataluren since the age of 2 years.
Exon skipping
Exon-skipping ASOs technology has the potential to induce cellular machinery to “skip over” a targeted exon and restore the reading frame, resulting in the production of internally truncated, but functional dystrophin protein [1]. Eteplirsen is a phosphorodiamidate morpholino oligomer which, delivered intravenously, targets the splice-donor region of exon 51 [33]. Eteplirsen, commercially available as Exondys 51™ in the USA, obtained in September 2016 a conditional approval by FDA to treat DMD patients amenable to exon 51 skipping (13% of DMD patients). The company supported its marketing request with results from two studies involving 12 boys with DMD, ages 7 to 13. The first, a phase 2 trial, randomized 12 patients to receive weekly intravenous infusions of eteplirsen or a placebo over 24 weeks. All study participants continued on weekly eteplirsen treatment into the second study, an open-label trial that ran for 3 years. The decline in the 6MWT was slower in the treated patients vs matched controls with a statistically significant difference of 151 m (p < 0.01). Moreover, the percentage of patients who lost the ambulation was lower in the treated patients vs matched controls (16.7% vs 46.2%) [34]. Furthermore, a clinically meaningful and statistically significant reduction in pulmonary function annual decline was demonstrated (2.3% vs 4.1%, respectively, in treated and natural-history controls) [35]. A confirmatory and open-label phase 3 study (NCT02255552), required by the FDA as part of its approval decision, is ongoing and due to conclude in May 2019.
In the European Union, the Committee for Medicinal Products for Human Use (CHMP), part of EMA, gave a negative opinion for eteplirsen in 2018. Sarepta Therapeutics, the company producing eteplirsen, appealed and a reevaluation started with a meeting involving also DMD experts and patient representatives. However, the second evaluation remained negative [36].
At the moment, Sarepta is developing a variety of other drug candidates based on proprietary RNA-based technology and unique phosphorodiamidate morpholino oligomer chemistry. A phase 3 trial is ongoing for golodirsen, able to skip exon 53 (8% of DMD patients), and casimersen for exon 45 skipping (8% of DMD patients) (NCT02500381). In December 2018, Wave Life Sciences Ltd., a biotechnology company focused on stereopure oligonucleotide skipping programme with higher efficacy than other ASOs [37], announced that the safety and tolerability data from the WVE-210201 (suvodirsen) phase 1 clinical trial in DMD boys who are amenable to exon 51 skipping support initiation of a phase 2/3 placebo-controlled efficacy and safety clinical trial in 2019 (https://ir.wavelifesciences.com/news-releases/news-release-details/wave-life-sciences-announces-positive-phase-1-results-wve-210201). Furthermore, several other therapeutic options are currently tested in experimental models and in DMD patients [38]. Table 1 summarizes the main therapeutic options currently tested in clinical trials.
X-linked myotubular myopathy
X-linked myotubular myopathy (XLMTM) is caused by mutations in the myotubularin gene (MTM1). The age of onset varies from the birth to young adulthood, and the clinical spectrum is variable ranging from individuals who require a wheelchair and full time breathing support to those who are able to walk and breathe on their own. Symptoms include a long face, facial and eye muscle weakness, hypotonia, and generalized muscle weakness. Muscle biopsy demonstrates central nucleated fibers, resembling myotubes [41]. Recently, the effects of MTM1 gene therapy using AAV8 vector on muscle weakness and pathology has been demonstrated in MTM1-mutant dogs with evident long-term benefits [42]. ASPIRO phase 1/2, multinational, open-label, ascending-dose, delayed-treatment concurrent control clinical study is ongoing to evaluate the safety and preliminary efficacy of a viral vector carrying the MTM1 gene (AT132) in subjects with XLMTM aged less than 5 years. Subjects receive a single dose of AT132 and are followed for safety and efficacy for 5 years (NCT03199469). An interim analysis presented at 2018 World Muscle Society Congress has reported increased limb and trunk strength, improved velocity and accuracy of movements, advanced ability to communicate, recovery in secretion management, and swallowing capability [43].
Non-dystrophic myotonias
Autosomal inherited non-dystrophic channelopathies include myotonia congenita (MC) and paramyotonia congenita (PMC), in which myotonia, disabling muscle stiffness and pain are the most troublesome symptoms that limit daily living [44]. MC exists in a dominantly (Thomsen myotonia) and a recessively inherited form (Becker myotonia), while all cases of PMC are dominantly inherited. In these conditions, myotonia usually presents in childhood and may affect all skeletal muscles, but the pattern and severity of muscle involvement varies among mutations, and individuals, even among family members. Mutations in the sarcolemmal chloride ion channel (CLCN1) reducing resting chloride conductance are the causes of MC, whereas mutations in the sodium ion channel gene (SCN4A) causing long-lasting depolarization due to impaired sodium conductance are associated with PMC [45]. In both MC and PMC, the changed conductance results in sarcolemmal hyperexcitability.
Several anticonvulsant drugs, antiarrhythmic drugs, and other channel blockers have been suggested as treatment for myotonia. The dramatic effect of flecainide on disabling muscle stiffness has been reported in anecdotical familial cases of MC linked to SCN4 mutations [46, 47]. However, mexiletine is to date the only drug that has shown evidence of alleviating myotonia [48]. Recently, lamotrigine has been investigated in a blinded, cross-over randomized placebo-controlled trial. The drug reduced self-assessed myotonia severity score and clinical myotonia and the SF-36 domain of physical function improved, with acceptable side effects [49]. More recently ranolazine, used to control chest pain in symptomatic coronary artery disease, which acts by enhancing slow inactivation of sodium channels, has been proposed as a therapeutic option in PMC and MC. Two open-label, single-center trials to evaluate efficacy and tolerability of ranolazine in patients with PMC and MC have been conducted in the last 2 years. Ranolazine was well tolerated without any serious adverse events, providing class IV evidence that ranolazine improves myotonia [50, 51].
Myotonic dystrophy type 1
Myotonic dystrophy type 1 (DM1) is the most common form of muscular dystrophy in adults, inherited with an autosomal dominant mechanism and caused by an unstable CTG expansion in the DM protein kinase (DMPK) gene. As a consequence, mutant transcripts containing expanded CUG repeats are retained in nuclear foci and alter the function of splicing regulatory factor members of the MBNL and CELF families, resulting in alternative splicing misregulation of specific transcripts in affected tissues. DM1 phenotype is characterized by a multisystemic array of symptoms, including distal and facial muscles wasting and weakness, myotonia, cataract, diabetes, hypogonadism, and arrhythmic cardiopathies [52].
In the recent years, it has been confirmed that the reduction of CUGexp RNA improves muscle strength in DM1 mice, suggesting that muscle weakness in DM1 patients may be improved following elimination of toxic RNAs [53]. A phase 1/2a blinded, placebo-controlled study to assess the safety, tolerability, and dose-range finding of multiple ascending doses of ISIS 598769, an ASO drug, was started with the aim to reduce the production of toxic DMPK RNA. The drug was administered subcutaneously to adult patients with DM1 with good security profile (NCT02312011).
Recently metformin, the most widely used antidiabetic drug, has been identified able to induce changes toward normalization in ratios of protein isoforms, the alteration of which is associated to various DM1 clinical symptoms [54]. A significant increase in 6MWT performance was identified in DM1 patients who fully completed a recent 52-week monocentric, double-blind, placebo-controlled phase 2 randomized study, suggesting promising effects of metformin on the mobility of patients [55].
Pompe disease
Pompe disease, a severe metabolic myopathy, is caused by mutations in the gene coding for acid alpha-glucosidase (GAA), the enzyme breaking down glycogen in the lysosomes. A deficiency of the enzyme leads to lysosomal accumulation of glycogen in multiple tissues, but cardiac and skeletal muscles are most severely affected. Pompe disease affects people of all ages with varying degrees of severity. The most severe form, referred to as classic infantile onset Pompe disease (IOPD), is characterized by the age of onset at ≤ 12 months, rapidly progressive hypertrophic cardiomyopathy, left ventricular outflow obstruction, hypotonia and muscle weakness, respiratory distress, and progressive loss of independent ventilation. Less severe is the late-onset form (LOPD) that manifests usually without significant cardiac involvement with symptoms resembling a proximal limb-girdle myopathy with respiratory failure due to the involvement of the diaphragm [56, 57].
Since 2006 the standard of care of Pompe disease is the enzyme replacement therapy (ERT). ERT dramatically changed the natural course of the disease in infants and resulted in much longer survival. The most reliable effect of ERT in infants has been demonstrated on cardiac pathology and motor functions as well as in adults, motor performances and respiratory parameters, although less impressively, were improved or maintained [58]. ERT efficacy depends on the mannose 6-phosphate (M6P) content of the recombinant human GAA (rhGAA) and on the abundance of the cation-independent mannose-6-phosphate receptor (CI-MPR) on the target tissue. M6P groups are critical for efficient uptake and lysosomal delivery of the recombinant enzyme. The limited effect of ERT in Pompe muscles has been mainly attributed to both low number of M6P groups on the rhGAA and the low expression of the receptor on the cell surface of muscle cells [59]. Several approaches designed to improve traditional ERT are currently under investigation. One of these is aimed at enhancing the enzyme delivery by increasing the number of M6P residues on the recombinant enzyme [60]. Neo-GAA (GZ402666, Sanofi Genzyme), a second-generation GAA with increased affinity for the CI-MPR, had well-tolerated safety profile and exploratory efficacy profile in a phase 1 study [61]. It is now tested in an ongoing phase 3 randomized, multicenter, multinational, double-blinded study (NCT02782741). The estimated primary completion date is February 2022.
An emerging strategy for the treatment of Pompe disease is chaperone therapy which relies on the ability of small-molecule pharmacological chaperones to promote folding, stability, and lysosomal trafficking of chaperone-responsive mutant enzymes [62]. In addition, the chaperones have been shown to have a stabilizing effect on the recombinant enzymes leading to their improved pharmacokinetics and pharmacodynamics. Indeed, improved stability of GAA in blood was observed in Pompe disease patients receiving ERT in combination with iminosugar N-butyldeoxynojirimycin [63]. A similar approach has been used by associating ERT with new investigational drug ATB200 alone or coadministered with the iminosugar miglustat. Safety, tolerability, and efficacy of coadministration are being investigated in a phase 2 clinical trial in LOPD (NCT02675465). Estimated primary completion date is September 2019.
A potential alternative to ERT is gene therapy. Systemic and intradiaphragmal delivery of rAAV1-hGAA was shown to improve respiratory function in KO mice. Subsequent studies demonstrated the capacity of AAV for retrograde movement and transduction of phrenic motoneurons. Based on these preclinical studies, the first-in-human trial of diaphragmatic gene therapy (AAV1-CMV-GAA) was conducted in children with IOPD who required assisted ventilation prior to the study. The safety of the AAV treatment was demonstrated but the clinical outcome was minimal. There was an anti-capsid and anti-transgene antibody response except for subjects who received concomitant immunomodulation [64, 65].
Another approach is liver-targeted gene therapy. To enhance systemic delivery and expression of GAA protein, investigators have harnessed the high metabolic capacity of the liver to produce and secrete the GAA protein. This strategy has been tried in GAA−/− mice and relies on infection with AAV8, which has a tropism for hepatic cells [66, 67]. Such a strategy could enhance and potentially replace ERT.
Mitochondrial DNA depletion induced by thymidine kinase 2 deficiency
Mitochondrial DNA (mtDNA) depletion syndrome is a frequent cause of severe childhood encephalomyopathy, associated to nuclear DNA gene mutation as thymidine kinase 2 (TK2). This gene encodes for a mitochondrial enzyme, involved into pyrimidine salvage pathway, that is essential for maintenance and replication of mtDNA. TK2 mutations can result in three disease subtypes: infantile-onset myopathy with rapid progression to early death; childhood-onset myopathy, similar to SMA type 3; and late-onset myopathy with moderate-to-severe manifestations as isolated chronic progressive external ophthalmoplegia (CPEO) or a generalized myopathy with CPEO, associated to facial and limb weakness [68]. Since TK2 deficiency results in the lack of deoxycytidine monophosphate (dCMP) and deoxythymidine monophosphate (dTMP), they were administered orally in TK2−/− mice, inducing a delayed onset of molecular and biochemical abnormalities, symptoms amelioration, and prolonged lifespan [69]. However, nucleotides were rapidly catabolized to nucleosides deoxycytidine (dC) and deoxythymidine (dT). To further enhance such therapy, direct dC and dT supplementation was recently tested in the mutant mice. This resulted in excellent restoring of mtDNA copy number and respiratory chain enzyme activities and levels, associated to a prolonged lifespan, supporting the use of this novel therapy in patients with TK2 deficiency [70].
Conclusions
In the past recent years, basic researchers and clinicians have worked together, often with the complementary and necessary help of advocacy groups, to guide research, to plan clinical trials, and to support requests to the regulatory agencies. A marvelous job has been done but still more work is ongoing to deal with new challenges. In SMA, which is the disease with the most innovative therapeutic advances, a previous consensus had already been decided upon a classification on current functional status in the form of non-sitters, sitters, and walkers [4, 5, 71]. Now, the changed clinical scenario because of therapeutic advances accentuates the need to plan a tailored care to single patient on the basis of the functional level, which will possibly change, for the first time, with a positive and not negative trajectory. Another important challenge in SMA is the newborn screening (NBS). The identification of therapeutic interventions that are more effective as early started and even more in the pre-symptomatic stage has created a pressing demand for NBS pilot studies to test the possibility to include SMA NBS in the public health programs [72].
Nusinersen, the first-ever therapy of SMA, has an extremely high cost, which has limited its market access in some countries. This issue has caused a large range of access type, from regular reimbursement to any SMA type to restrictions to type, age, or further inclusion/exclusion clinical criteria established by national committees. The high social and economic burden of SMA had been already discussed [73, 74], and the high price of nusinersen has stimulated pharmacoeconomics and cost-effectiveness ratio analyses [75]. On the other hand, the absence of any drug for SMA for decades has caused very high patient’s and caregiver’s expectations for nusinersen [76, 77], despite the different level of functional improvement obtainable according to time of treatment start [12]. Presumably, we are just at the beginning of a very wide discussion about the cost of innovative drugs for rare diseases and specific cost-benefit analysis, which has to consider all direct and indirect effects, real and perceived, on patients, their family, and society.
Achievements in new successful therapeutic strategies in one disease can produce further impetus on research approaches in other diseases. This already occurred after the first development of ASOs and gene therapy. The “new frontier” is now CRISPR-Cas9 genome editing system to cut and repair a specific target sequence of DNA in a genome. This approach was tried to rescue RNA toxicity in myoblasts from transgenic DM1 mouse model and DM1 patients. Myogenic capacity, nucleocytoplasmic distribution, and abnormal ribonucleoprotein particle-binding behaviour of transcripts from the edited DMPK gene were normalized, suggesting new therapeutic opportunities for DM1 patients [78]. In a very recent experiment, AAVs were used to deliver CRISPR gene editing components to canine DMD model by intramuscular delivery. Dystrophin protein expression was restored to levels ranging from 3 to 90% of normal, depending on muscle type. In cardiac muscle, dystrophin levels reached 92% of normal. Both skeletal and cardiac muscles showed improved muscle histology [79]. Finally, multiple therapeutic approaches described here, including already commercially available drugs, just approved drugs and new therapeutic promises, give us good reasons to be optimistic about the future.
References
Li D, Mastaglia FL, Fletcher S, Wilton SD (2018) Precision medicine through antisense oligonucleotide-mediated exon skipping. Trends Pharmacol Sci 39:982–994
Quinlivan R, Matthews E, Hanna MG (2014) Innovative care model for patients with complex muscle diseases. Curr Opin Neurol 27:607–613
Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Colvin MK, Cripe L, Herron AR, Kennedy A, Kinnett K, Naprawa J, Noritz G, Poysky J, Street N, Trout CJ, Weber DR, Ward LM, DMD Care Considerations Working Group (2018) Diagnosis and management of Duchenne muscular dystrophy, part 3: primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol 17:445–455
Finkel RS, Mercuri E, Meyer OH, Simonds AK, Schroth MK, Graham RJ, Kirschner J, Iannaccone ST, Crawford TO, Woods S, Muntoni F, Wirth B, Montes J, Main M, Mazzone ES, Vitale M, Snyder B, Quijano-Roy S, Bertini E, Davis RH, Qian Y, Sejersen T, SMA Care group (2018) Diagnosis and management of spinal muscular atrophy: part 2: pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord 28:197–207
Mercuri E, Finkel RS, Muntoni F, Wirth B, Montes J, Main M, Mazzone ES, Vitale M, Snyder B, Quijano-Roy S, Bertini E, Davis RH, Meyer OH, Simonds AK, Schroth MK, Graham RJ, Kirschner J, Iannaccone ST, Crawford TO, Woods S, Qian Y, Sejersen T, SMA Care Group (2018) Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord 28:103–115
Calucho M, Bernal S, Alías L, March F, Venceslá A, Rodríguez-Álvarez FJ, Aller E, Fernández RM, Borrego S, Millán JM, Hernández-Chico C, Cuscó I, Fuentes-Prior P, Tizzano EF (2018) Correlation between SMA type and SMN2 copy number revisited: an analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord 28:208–215
Messina S (2018) New directions for SMA therapy. J Clin Med 7(9)
Bertini E, Dessaud E, Mercuri E, Muntoni F, Kirschner J, Reid C, Lusakowska A, Comi GP, Cuisset JM, Abitbol JL, Scherrer B, Ducray PS, Buchbjerg J, Vianna E, van der Pol WL, Vuillerot C, Blaettler T, Fontoura P, Olesoxime SMA Phase 2 Study Investigators (2017) Safety and efficacy of olesoxime in patients with type 2 or non-ambulatory type 3 spinal muscular atrophy: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol 16:513–522
Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, Chiriboga CA, Saito K, Servais L, Tizzano E, Topaloglu H, Tulinius M, Montes J, Glanzman AM, Bishop K, Zhong ZJ, Gheuens S, Bennett CF, Schneider E, Farwell W, De Vivo DC, ENDEAR Study Group (2017) Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med 377:1723–1732
Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, Iannaccone ST, Kirschner J, Kuntz NL, Saito K, Shieh PB, Tulinius M, Mazzone ES, Montes J, Bishop KM, Yang Q, Foster R, Gheuens S, Bennett CF, Farwell W, Schneider E, De Vivo DC, Finkel RS, CHERISH Study Group (2018) Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med 378:625–635
Messina S, Pane M, Sansone V, Bruno C, Catteruccia M, Vita G, Palermo C, Albamonte E, Pedemonte M, Bertini E, Binetti L, Mercuri E, Italian EAP working Group (2017) Expanded access program with nusinersen in SMA type I in Italy: strengths and pitfalls of a successful experience. Neuromuscul Disord 27:1084–1086
Pane M, Palermo C, Messina S, Sansone VA, Bruno C, Catteruccia M, Sframeli M, Albamonte E, Pedemonte M, D’Amico A, Brigati G, de Sanctis R, Coratti G, Lucibello S, Bertini E, Vita G, Tiziano FD, Mercuri E, Italian EAP working group (2018) Nusinersen in type 1 SMA infants, children and young adults: preliminary results on motor function. Neuromuscul Disord 28:582–585
New warning of nusinersen-related communicating hydrocephalus (2018) Reactions Weekly 1714:3. https://doi.org/10.1007/s40278-018-50183-2
Ratni H, Ebeling M, Baird J, Bendels S, Bylund J, Chen KS, Denk N, Feng Z, Green L, Guerard M, Jablonski P, Jacobsen B, Khwaja O, Kletzl H, Ko CP, Kustermann S, Marquet A, Metzger F, Mueller B, Naryshkin NA, Paushkin SV, Pinard E, Poirier A, Reutlinger M, Weetall M, Zeller A, Zhao X, Mueller L (2018) Discovery of risdiplam, a selective survival of motor neuron-2 ( SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J Med Chem 61:6501–6517
Sturm S, Günther A, Jaber B, Jordan P, Al Kotbi N, Parkar N, Cleary Y, Frances N, Bergauer T, Heinig K, Kletzl H, Marquet A, Ratni H, Poirier A, Müller L, Czech C, Khwaja O (2019) A phase 1 healthy male volunteer single escalating dose study of the pharmacokinetics and pharmacodynamics of risdiplam (RG7916, RO7034067), a SMN2 splicing modifier. Br J Clin Pharmacol 85:181–193
Meyer K, Ferraiuolo L, Schmelzer L, Braun L, McGovern V, Likhite S, Michels O, Govoni A, Fitzgerald J, Morales P, Foust KD, Mendell JR, Burghes AH, Kaspar BK (2015) Improving single injection CSF delivery of AAV9-mediated gene therapy for SMA: a dose-response study in mice and nonhuman primates. Mol Ther 23:477–487
Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, Lowes L, Alfano L, Berry K, Church K, Kissel JT, Nagendran S, L'Italien J, Sproule DM, Wells C, Cardenas JA, Heitzer MD, Kaspar A, Corcoran S, Braun L, Likhite S, Miranda C, Meyer K, Foust KD, Burghes AHM, Kaspar BK (2017) Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med 377:1713–1722
Al-Zaidy S, Pickard AS, Kotha K, Alfano LN, Lowes L, Paul G, Church K, Lehman K, Sproule DM, Dabbous O, Maru B, Berry K, Arnold WD, Kissel JT, Mendell JR, Shell R (2019) Health outcomes in spinal muscular atrophy type 1 following AVXS-101 gene replacement therapy. Pediatr Pulmonol 54:179–185
Messina S, Bitto A, Aguennouz M, Mazzeo A, Migliorato A, Polito F, Irrera N, Altavilla D, Vita GL, Russo M, Naro A, De Pasquale MG, Rizzuto E, Musarò A, Squadrito F, Vita G (2009) Flavocoxid counteracts muscle necrosis and improves functional properties in mdx mice: a comparison study with methylprednisolone. Exp Neurol 220:349–358
Messina S, Bitto A, Aguennouz M, Vita GL, Polito F, Irrera N, Altavilla D, Marini H, Migliorato A, Squadrito F, Vita G (2011) The soy isoflavone genistein blunts nuclear factor kappa-B, MAPKs and TNF-α activation and ameliorates muscle function and morphology in mdx mice. Neuromuscul Disord 21:579–589
Messina S, Bitto A, Vita GL, Aguennouz M, Irrera N, Licata N, Sframeli M, Bruschetta D, Minutoli L, Altavilla D, Vita G, Squadrito F (2015) Modulation of neuronal nitric oxide synthase and apoptosis by the isoflavone genistein in mdx mice. BioFactors 41:324–329
Vita GL, Polito F, Oteri R, Arrigo R, Ciranni AM, Musumeci O, Messina S, Rodolico C, Di Giorgio RM, Vita G, Aguennouz M (2018) Hippo signaling pathway is altered in Duchenne muscular dystrophy. PLoS One 13(10):e0205514
D'Amico A, Catteruccia M, Baranello G, Politano L, Govoni A, Previtali SC, Pane M, D'Angelo MG, Bruno C, Messina S, Ricci F, Pegoraro E, Pini A, Berardinelli A, Gorni K, Battini R, Vita G, Trucco F, Scutifero M, Petillo R, D’Ambrosio P, Ardissone A, Pasanisi B, Vita G, Mongini T, Moggio M, Comi GP, Mercuri E, Bertini E (2017) Diagnosis of Duchenne muscular dystrophy in Italy in the last decade: critical issues and areas for improvements. Neuromuscul Disord 27:447–451
Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, Case LE, Clemens PR, Hadjiyannakis S, Pandya S, Street N, Tomezsko J, Wagner KR, Ward LM, Weber DR, DMD Care Considerations Working Group (2018) Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol 17:251–267
Birnkrant DJ, Bushby K, Bann CM, Alman BA, Apkon SD, Blackwell A, Case LE, Cripe L, Hadjiyannakis S, Olson AK, Sheehan DW, Bolen J, Weber DR, Ward LM, DMD Care Considerations Working Group (2018) Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol 17:347–361
Rahbek J, Steffensen BF, Bushby K, de Groot IJ (2015) 206th ENMC international Workshop: care for a novel group of patients - adults with Duchenne muscular dystrophy. Naarden, The Netherlands, 23-25 May 2014. Neuromuscul Disord 25:727–738
Bach JR, Martinez D (2011) Duchenne muscular dystrophy: continuous noninvasive ventilatory support prolongs survival. Respir Care 56:744–750
Messina S, Vita GL (2018) Clinical management of Duchenne muscular dystrophy: the state of the art. Neurol Sci 39:1837–1845. https://doi.org/10.1007/s10072-018-3555-3
Matthews E, Brassington R, Kuntzer T, Jichi F, Manzur AY (2016) Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst Rev (5):CD003725
Guglieri M, Bushby K, McDermott MP, Hart KA, Tawil R, Martens WB, Herr BE, McColl E, Wilkinson J, Kirschner J, King WM, Eagle M, Brown MW, Willis T, Hirtz D, Shieh PB, Straub V, Childs AM, Ciafaloni E, Butterfield RJ, Horrocks I, Spinty S, Flanigan KM, Kuntz NL, Baranello G, Roper H, Morrison L, Mah JK, Manzur AY, McDonald CM, Schara U, von der Hagen M, Barohn RJ, Campbell C, Darras BT, Finkel RS, Vita G, Hughes I, Mongini T, Pegoraro E, Wicklund M, Wilichowski E, Bryan Burnette W, Howard JF, McMillan HJ, Thangarajh M, Griggs RC (2017) Developing standardized corticosteroid treatment for Duchenne muscular dystrophy. Contemp Clin Trials 58:34–39
Mah JK (2018) An overview of recent therapeutics advances for Duchenne muscular dystrophy. Methods Mol Biol 1687:3–17
McDonald CM, Campbell C, Torricelli RE, Finkel RS, Flanigan KM, Goemans N, Heydemann P, Kaminska A, Kirschner J, Muntoni F, Osorio AN, Schara U, Sejersen T, Shieh PB, Sweeney HL, Topaloglu H, Tulinius M, Vilchez JJ, Voit T, Wong B, Elfring G, Kroger H, Luo X, McIntosh J, Ong T, Riebling P, Souza M, Spiegel RJ, Peltz SW, Mercuri E, Clinical Evaluator Training Group, ACT DMD Study Group (2017) Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390:1489–1498
Stein CA, Castanotto D (2017) FDA-approved oligonucleotide therapies in 2017. Mol Ther 25:1069–1075
Mendell JR, Goemans N, Lowes LP, Alfano LN, Berry K, Shao J, Kaye EM, Mercuri E, Eteplirsen Study Group and Telethon Foundation DMD Italian Network (2016) Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol 79:257–271
Kinane TB, Mayer OH, Duda PW, Lowes LP, Moody SL, Mendell JR (2018) Long-term pulmonary function in Duchenne muscular dystrophy: comparison of eteplirsen-treated patients to natural history. J Neuromuscul Dis 5:47–58
Aartsma-Rus A, Goemans N (2019) A sequel to the eteplirsen saga: eteplirsen is approved in the United States but was not approved in Europe. Nucleic Acid Ther 29:13–15
Iwamoto N, Butler DCD, Svrzikapa N, Mohapatra S, Zlatev I, Sah DWY, Meena SSM, Lu G, Apponi LH, Frank-Kamenetsky M, Zhang JJ, Vargeese C, Verdine GL (2017) Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat Biotechnol 35:845–851
Barthélémy F, Wein N (2018) Personalized gene and cell therapy for Duchenne muscular dystrophy. Neuromuscul Disord 28:803–824
Bettica P, Petrini S, D'Oria V, D'Amico A, Catteruccia M, Pane M, Sivo S, Magri F, Brajkovic S, Messina S, Vita GL, Gatti B, Moggio M, Puri PL, Rocchetti M, De Nicolao G, Vita G, Comi GP, Bertini E, Mercuri E (2016) Histological effects of givinostat in boys with Duchenne muscular dystrophy. Neuromuscul Disord 26:643–649
Hoffman EP, Riddle V, Siegler MA, Dickerson D, Backonja M, Kramer WG, Nagaraju K, Gordish-Dressman H, Damsker JM, McCall JM (2018) Phase 1 trial of vamorolone, a first-in-class steroid, shows improvements in side effects via biomarkers bridged to clinical outcomes. Steroids 134:43–52
Cassandrini D, Trovato R, Rubegni A, Lenzi S, Fiorillo C, Baldacci J, Minetti C, Astrea G, Bruno C, Santorelli FM, Italian Network on Congenital Myopathies (2017) Congenital myopathies: clinical phenotypes and new diagnostic tools. Ital J Pediatr 43:101
Elverman M, Goddard MA, Mack D, Snyder JM, Lawlor MW, Meng H, Beggs AH, Buj-Bello A, Poulard K, Marsh AP, Grange RW, Kelly VE, Childers MK (2017) Long-term effects of systemic gene therapy in a canine model of myotubular myopathy. Muscle Nerve 56:943–953
Kuntz N, Shieh P, Smith B, Bönnemann C, Dowling J, Lawlor M, Müller-Felber W, Noursalehi M, Rico S, Servais L, Prasad S (2018) ASPIRO phase 1/2 gene therapy trial in X-linked myotubular myopathy: preliminary safety and efficacy findings. Neuromuscul Disord 28(Supp. 2):S91
Trivedi JR, Bundy B, Statland J, Salajegheh M, Rayan DR, Venance SL, Wang Y, Fialho D, Matthews E, Cleland J, Gorham N, Herbelin L, Cannon S, Amato A, Griggs RC, Hanna MG, Barohn RJ, CINCH Consortium (2013) Non-dystrophic myotonia: prospective study of objective and patient reported outcomes. Brain 136:2189–2200
Matthews E, Fialho D, Tan SV, Venance SL, Cannon SC, Sternberg D, Fontaine B, Amato AA, Barohn RJ, Griggs RC, Hanna MG, Investigators CINCH (2010) The non-dystrophic myotonias: molecular pathogenesis, diagnosis and treatment. Brain 133:9–22
Portaro S, Rodolico C, Sinicropi S, Musumeci O, Valenzise M, Toscano A (2016) Flecainide-responsive myotonia permanens with SNEL onset: a new case and literature review. Pediatrics 137:e20153289
Terracciano C, Farina O, Esposito T, Lombardi L, Napolitano F, Blasiis P, Ciccone G, Todisco V, Tuccillo F, Bernardini S, Di Iorio G, Melone MAB, Sampaolo S (2018) Successful long-term therapy with flecainide in a family with paramyotonia congenita. J Neurol Neurosurg Psychiatry 89:1232–1234
Statland J, Bundy BN, Wang Y, Rayan DR, Trivedi JR, Sansone VA, Salajegheh MK, Venance SL, Ciafaloni E, Matthews E, Meola G, Herbelin L, Griggs RC, Barohn RJ, Hanna MG, Consortium for Clinical Investigation of Neurologic Channelopathies (2012) Mexiletine for symptoms and signs of myotonia in nondystrophic myotonia: a randomized controlled trial. JAMA 308:1357–1365
Andersen G, Hedermann G, Witting N, Duno M, Andersen H, Vissing J (2017) The antimyotonic effect of lamotrigine in non-dystrophic myotonias: a double-blind randomized study. Brain 140:2295–2305
Arnold WD, Kline D, Sanderson A, Hawash AA, Bartlett A, Novak KR, Rich MM, Kissel JT (2017) Open-label trial of ranolazine for the treatment of myotonia congenita. Neurology 89:710–713
Lorusso S, Kline D, Bartlett A, Freimer M, Agriesti J, Hawash AA, Rich MM, Kissel JT, David Arnold W (2019) Open-label trial of ranolazine for the treatment of paramyotonia congenita. Muscle Nerve 59:240–243
De Antonio M, Dogan C, Hamroun D, Mati M, Zerrouki S, Eymard B, Katsahian S, Bassez G, French Myotonic Dystrophy Clinical Network (2016) Unravelling the myotonic dystrophy type 1 clinical spectrum: a systematic registry-based study with implications for disease classification. Rev Neurol (Paris) 172:572–580
Jauvin D, Chrétien J, Pandey SK, Martineau L, Revillod L, Bassez G, Lachon A, MacLeod AR, Gourdon G, Wheeler TM, Thornton CA, Bennett CF, Puymirat J (2017) Targeting DMPK with antisense oligonucleotide improves muscle strength in myotonic dystrophy type 1 mice. Mol Ther Nucleic Acids 7:465–474
Laustriat D, Gide J, Barrault L, Chautard E, Benoit C, Auboeuf D, Boland A, Battail C, Artiguenave F, Deleuze JF, Bénit P, Rustin P, Franc S, Charpentier G, Furling D, Bassez G, Nissan X, Martinat C, Peschanski M, Baghdoyan S (2015) In vitro and in vivo modulation of alternative splicing by the biguanide metformin. Mol Ther Nucleic Acids 4:e262
Bassez G, Audureau E, Hogrel JY, Arrouasse R, Baghdoyan S, Bhugaloo H, Gourlay-Chu ML, Le Corvoisier P, Peschanski M (2018) Improved mobility with metformin in patients with myotonic dystrophy type 1: a randomized controlled trial. Brain 141:2855–2865
Van der Ploeg AT, Reuser AJ (2008) Pompe’s disease. Lancet 372:1342–1353
Angelini C, Semplicini C, Ravaglia S, Bembi B, Servidei S, Pegoraro E, Moggio M, Filosto M, Sette E, Crescimanno G, Tonin P, Parini R, Morandi L, Marrosu G, Greco G, Musumeci O, Di Iorio G, Siciliano G, Donati MA, Carubbi F, Ermani M, Mongini T, Toscano A, Italian GSDII Group (2012) Observational clinical study in juvenile-adult glycogenosis type 2 patients undergoing enzyme replacement therapy for up to 4 years. J Neurol 259:952–958
Schoser B, Stewart A, Kanters S, Hamed A, Jansen J, Chan K, Karamouzian M, Toscano A (2017) Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. J Neurol 264:621–630
Koeberl DD, Luo X, Sun B, McVie-Wylie A, Dai J, Li S, Banugaria SG, Chen YT, Bali DS (2011) Enhanced efficacy of enzyme replacement therapy in Pompe disease through mannose-6-phosphate receptor expression in skeletal muscle. Mol Genet Metab 103:107–112
Maga JA, Zhou J, Kambampati R, Peng S, Wang X, Bohnsack RN, Thomm A, Golata S, Tom P, Dahms NM, Byrne BJ, LeBowitz JH (2013) Glycosylation independent lysosomal targeting of acid alpha-glucosidase enhances muscle glycogen clearance in Pompe mice. J Biol Chem 288:1428–1438
Pena LD, Barohn RJ, Byrne BJ, Desnuelle C, Goker-Alpan O, Ladha S, Laforêt P, Mengel KE, Pestronk A, Pouget J, Schoser B, Straub V, Trivedi J, Van Damme P, Vissing J, Young P, Kacena K, Shafi R, Thurberg BL, Culm-Merdek K, van der Ploeg AT, on behalf of the NEO1 Investigator Group (2018) Safety, tolerability, pharmacokinetics, pharmacodynamics, and exploratory efficacy of the novel enzyme replacement therapy avalglucosidase alfa (neoGAA) in treatment-naïve and alglucosidase alfa-treated patients with late-onset Pompe disease: a phase 1, open-label, multicenter, multinational, ascending dose study. Neuromuscul Disord. Dec 17. https://doi.org/10.1016/j.nmd.2018.12.004
Parenti G, Moracci M, Fecarotta S, Andria G (2014) Pharmacological chaperone therapy for lysosomal storage diseases. Future Med Chem 6:1031–1045
Parenti G, Fecarotta S, la Marca G, Rossi B, Ascione S, Donati MA, Morandi LO, Ravaglia S, Pichiecchio A, Ombrone D, Sacchini M, Pasanisi MB, De Filippi P, Danesino C, Della Casa R, Romano A, Mollica C, Rosa M, Agovino T, Nusco E, Porto C, Andria G (2014) A chaperone enhances blood α-glucosidase activity in Pompe disease patients treated with enzyme replacement therapy. Mol Ther 22:2004–2012
Smith BK, Martin AD, Lawson LA, Vernot V, Marcus J, Islam S, Shafi N, Corti M, Collins SW, Byrne BJ (2017) Inspiratory muscle conditioning exercise and diaphragm gene therapy in Pompe disease: clinical evidence of respiratory plasticity. Exp Neurol 287:216–224
Corti M, Liberati C, Smith BK, Lawson LA, Tuna IS, Conlon TJ, Coleman KE, Islam S, Herzog RW, Fuller DD, Collins SW, Byrne BJ (2017) Safety of intradiaphragmatic delivery of adeno-associated virus-mediated alpha-glucosidase (rAAV1-CMV-hGAA) gene therapy in children affected by Pompe disease. Hum Gene Ther Clin Dev 28:208–218
Han SO, Ronzitti G, Arnson B, Leborgne C, Li S, Mingozzi F, Koeberl D (2017) Low-dose liver-targeted gene therapy for Pompe disease enhances therapeutic efficacy of ERT via immune tolerance induction. Mol Ther Methods Clin Dev 4:126–136
Puzzo F, Colella P, Biferi MG, Bali D, Paulk NK, Vidal P, Collaud F, Simon-Sola M, Charles S, Hardet R, Leborgne C, Meliani A, Cohen-Tannoudji M, Astord S, Gjata B, Sellier P, van Wittenberghe L, Vignaud A, Boisgerault F, Barkats M, Laforet P, Kay MA, Koeberl DD, Ronzitti G, Mingozzi F (2017) Rescue of Pompe disease in mice by AAV-mediated liver delivery of secretable acid α-glucosidase. Sci Transl Med 9(418):eaam6375
Garone C, Taylor RW, Nascimento A, Poulton J, Fratter C, Domínguez-González C, Evans JC, Loos M, Isohanni P, Suomalainen A, Ram D, Hughes MI, McFarland R, Barca E, Lopez Gomez C, Jayawant S, Thomas ND, Manzur AY, Kleinsteuber K, Martin MA, Kerr T, Gorman GS, Sommerville EW, Chinnery PF, Hofer M, Karch C, Ralph J, Cámara Y, Madruga-Garrido M, Domínguez-Carral J, Ortez C, Emperador S, Montoya J, Chakrapani A, Kriger JF, Schoenaker R, Levin B, Thompson JLP, Long Y, Rahman S, Donati MA, DiMauro S, Hirano M (2018) Retrospective natural history of thymidine kinase 2 deficiency. J Med Genet 55:515–521
Garone C, Garcia-Diaz B, Emmanuele V, Lopez LC, Tadesse S, Akman HO, Tanji K, Quinzii CM, Hirano M (2014) Deoxypyrimidine monophosphate bypass therapy for thymidine kinase 2 deficiency. EMBO Mol Med 6:1016–1027
Lopez-Gomez C, Levy RJ, Sanchez-Quintero MJ, Juanola-Falgarona M, Barca E, Garcia-Diaz B, Tadesse S, Garone C, Hirano M (2017) Deoxycytidine and deoxythymidine treatment for thymidine kinase 2 deficiency. Ann Neurol 81:641–652
Finkel RS, Sejersen T, Mercuri E, ENMC SMA Workshop Study Group (2016) 218th ENMC International Workshop: Revisiting the consensus on standards of care in SMA. Naarden, The Netherlands, 19–21 February 2016. Neuromuscul Disord 27:596–605
Tizzano EF, Zafeiriou D (2018) Prenatal aspects in spinal muscular atrophy: from early detection to early presymptomatic intervention. Eur J Paediatr Neurol 22:944–950
Klug C, Schreiber-Katz O, Thiele S, Schorling E, Zowe J, Reilich P, Walter MC, Nagels KH (2016) Disease burden of spinal muscular atrophy in Germany. Orphanet J Rare Dis 11(1):58
López-Bastida J, Peña-Longobardo LM, Aranda-Reneo I, Tizzano E, Sefton M, Oliva-Moreno J (2017) Social/economic costs and health-related quality of life in patients with spinal muscular atrophy (SMA) in Spain. Orphanet J Rare Dis 12(1):141
Zuluaga-Sanchez S, Teynor M, Knight C, Thompson R, Lundqvist T, Ekelund M, Forsmark A, Vickers AD, Lloyd A (2019) Cost effectiveness of nusinersen in the treatment of patients with infantile-onset and later-onset spinal muscular atrophy in Sweden. Pharmacoeconomics Feb 4. https://doi.org/10.1007/s40273-019-00769-6
La Foresta S, Faraone C, Sframeli M, Vita GL, Russo M, Profazio C, Rulli I, Gitto E, Versaci A, Messina S, Vita G (2018) Intrathecal administration of nusinersen in type 1 SMA: successful psychological program in a single Italian center. Neurol Sci 39:1961–1964
Pacione M, Siskind CE, Day JW, Tabor HK (2019) Perspectives on Spinraza (nusinersen) treatment study: views of individuals and parents of children diagnosed with spinal muscular atrophy. J Neuromuscul Dis 6:119–131
van Agtmaal EL, André LM, Willemse M, Cumming SA, van Kessel IDG, van den Broek WJAA, Gourdon G, Furling D, Mouly V, Monckton DG, Wansink DG, Wieringa B (2017) CRISPR/Cas9-induced (CTG·CAG)n repeat instability in the myotonic dystrophy type 1 locus: implications for therapeutic genome editing. Mol Ther 25:24–43
Amoasii L, Hildyard JCW, Li H, Sanchez-Ortiz E, Mireault A, Caballero D, Harron R, Stathopoulou TR, Massey C, Shelton JM, Bassel-Duby R, Piercy RJ, Olson EN (2018) Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 362:86–91
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
G.V. discloses having been on advisory board for Avexis and Roche. He is also a principal investigator in clinical trials sponsored by Avexis, Roche, Sarepta, Santhera, Italfarmaco, Wave, and PTC.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Vita, G., Vita, G.L., Musumeci, O. et al. Genetic neuromuscular disorders: living the era of a therapeutic revolution. Part 2: diseases of motor neuron and skeletal muscle. Neurol Sci 40, 671–681 (2019). https://doi.org/10.1007/s10072-019-03764-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-019-03764-z