
Abstract
Traumatic brain injury is one of the most common causes for intervention in neurosurgery. Apart from its acute consequence, it can represent a further burden on individuals as well as society by being associated with significant comorbidity—mainly early-onset dementia. Oxidative stress is one of the crucial mechanisms conferring the damage to nervous tissue, and it is believed it could be, to some extent, influenced by dietary composition, largely by antioxidants contained in the diet. Under stressful conditions, cell-derived reactive oxygen species in the brain can induce the formation of lipid peroxides and the shifting of redox homeostasis. This review discusses the potential of vitamin E as a potent antioxidant and its derived molecules, including vitamin E-based lazaroids, in traumatic brain injury, summarizing the current state of knowledge of its role in TBI-associated dementia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Traumatic brain injury (TBI) is one of the leading causes requiring neurosurgical intervention and is characterized by a primary penetrating or non-penetrating injury of the head, or as a secondary injury linked to subsequent maladaptive reactions of the body. Epidemiological evidence suggests approximately 80% of TBI cases are mild, of which a considerable proportion of cases go unrecognized in the community [1]. TBI is recognized as a contributing factor in cognition deterioration and dementia, and its relation to dementia have been studied for many years [2]. It has been described that a history of TBI correlates with elevated risk for developing cognitive impairment and dementia, either the Alzheimer’s disease (AD) type or the non-AD type [3, 4]. It is also known that the severity of TBI correlates positively with the risk of cognitive impairment/dementia development. In a recent study, it was also reported that a history of TBI may accelerate the age-of-onset of cognitive impairment/dementia by two or more years [2].
In the European Union, brain injury accounts for one million hospital admissions per year, representing an enormous socio-economic burden for individuals, as well as society as a whole [5]. In a large review of TBI-related articles from about 1980 to 2003 performed by Tagliaferri et al., an aggregate hospitalized plus fatal TBI incidence rate of about 235 per 100,000 was estimated [6]. A more recent meta-analysis of 28 epidemiological studies on TBI from 16 European countries reveals an estimated overall incidence rate of 262 per 100,000 for admitted TBI, suggesting either more cases of TBI or that milder forms are more likely to be detected by healthcare systems [7]. While Tagliaferri et al. reported the main cause of TBI to be road traffic accidents (RTAs), Peeters et al. [7], on the other hand, report falls to be the most common cause. Meta-analysis by Peeters et al. fails to indicate any trends towards decreasing prevalence/incidence of TBI in Europe compared to 2006, despite the fact it reports a lower mortality rate than that in the study by Tagliaferri et al. Both reviews, however, emphasize the necessity for the implementation of unified guidelines, as well as the need for generalized/standardized case definitions, case ascertainment and treatment methods. While potential treatment methods for acute TBI are subject with much attention from physicians and researchers alike, the long-term adverse consequences of TBI have been far less investigated. In particular, very little is currently known about possible ways of modulating the pathological cascade in TBI, especially preventing oxidative damage and its consequences.
The aim of this review is, therefore, to summarize current knowledge on the localized activity of vitamin E as an important free radical scavenger in the brain and its possible prognostic benefits in TBI.
Traumatic brain injury
Traumatic brain injury (TBI) is a non-degenerative insult to the brain from an external mechanical force, leading to permanent or temporary impairment of cognitive, physical and psychosocial functions, associated with diminished or an altered state of consciousness. TBI severity classification is traditionally made using the Glasgow Coma Scale (GCS), with scores upon hospital admission ranging from mild (GCS 13–15), moderate (GCS 9–12) to severe (GCS ≤ 8). The actual value of GCS also reflects the risk of dying from TBI, which is low after mild (∼ 1%), intermediate after moderate (up to 15%), and high (up to 40%) after severe cases of TBI [8]. Although prognosis is often predicted based on measured clinical severity, the extent to which each of these severity assessments correlates with the outcome is less clear [9]. In 1996, the Brain Trauma Foundation (BTF) published the first guidelines on the management of severe TBI [10], accepted by the American Association of Neurological Surgeons, further approved and accepted by the World Health Organization Committee on Neurotrauma [11].
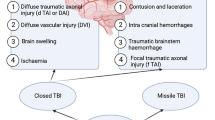
TBI can present itself both as a diffuse injury associated with diffuse axonal injury (DAI) and brain oedema, or as focal TBI following a direct impact to the head [8]. Although a significant segment of injuries may be considered predominantly focal or diffuse, in most cases, histopathology of the diseases is heterogeneous viz. both focal and diffuse components [9]. It has, in addition, been reported that the mortality rate for severe focal injuries is approximately 40%, and for severe diffuse injuries, approximately 25% [5].
On the basis of their particular involvement with the cascade of pathophysiological mechanisms, TBI can be also characterized as primary or secondary phase, both contributing in their own way to the extent of damage [12]. Even though the progressive character of the pathology and its long-term negative outcomes after TBI are generally recognized, scarce studies focus on the exact mechanisms of TBI damage. Primary injury after TBI results from external mechanical forces causing damage to brain tissue with probable brain malfunction. Among these forces are accelerating and decelerating linear forces, rotational forces, blunt impact, penetrating projectile and blast injury. All these forces are capable of causing direct damage to neurons, axons, dendrites, glial cell and vasculature across the spectrum of focal, multifocal or diffuse patterns. Once the trauma occurs, the consequences of primary damage are usually unavoidable and cannot be massively altered. Secondary damage results, on the contrary, from the complications of primary brain injury, often associated with brain oedema, increased intracranial pressure and opportunistic infections. Secondary brain injury is further associated with a multitude of pathogenic mechanisms, mainly mitochondrial dysfunction, diffuse axonal damage, inflammation, excitotoxicity and oxidative damage [12].
Depending on its severity, TBI is generally associated with significant deficits of cognitive executive function, including memory, planning, judgment, decision-making and the top-down control of emotions and behavioural executive functions, such as the emotional control component of decision-making, motivation and impulsivity [13]. Depending upon the severity of primary injury and its associated secondary phase, damage can be deemed transient, short-term, long-term or permanent. In a survey of very long-term outcomes of TBI sequelae, memory impairments were reported most frequently [14]. Unlike in amnestic memory disorders, memory problems in TBI are not typically due to a deficit of memory storage, meaning that TBI patients hold their ability to recognize the newly learned material; however, they tend to have difficulties organizing new information for successful encoding and retrieval [13]. Most frequently, the memory deficit presents itself first as post-traumatic amnesia, subsequently improving upon recovery [15]. The aetiology of the cognitive dysfunction is highly complex and for the greater part, remains a conundrum.
Risk of dementia in TBI
The mechanisms underlying the relationship between TBI and subsequent development of dementia are unclear; however, empirical observations support the hypothesis of higher prevalence of dementia among TBI patients. Apart from the clear epidemiological findings, TBI has also been associated with distinct histopathological findings typical for dementia, such as diffuse brain atrophy, cavum septum pellucidum, and amyloid-β (Aβ), tau and TDP-43 pathologies [16]. In a key study from 2014, looking at 164,661 hospitalized patients who suffered either TBI or non-TBI, a total of 51,799 patients with trauma (31.5%) were identified as TBI patients. Of these TBI cases, 4361 (8.4%) developed dementia, compared with 6610 patients in the non-TBI subset (5.9%) [17]. One hypothesis has been proposed that mild TBI predisposes to chronic traumatic encephalopathy (CTE), while moderate to severe TBI increases the risk of developing Alzheimer’s disease (AD) later in life [16].
Besides the gross morphological pathologies in severe TBI, essentially brain atrophy, cavum septum pellucidum and visible ventricular enlargement, more subtle processes can be observed at the histological level. The patients presenting with milder TBI, for instance pathologies, tauopathies and TAR DNA-binding protein aberrations, neuronal loss, changes in substantia nigra and white matter degeneration, have all been previously associated with dementia development [16].
In an immunohistochemical study that investigated the extent of AD-related changes in temporal lobe cortical biopsy resected from individuals treated surgically for severe TBI, diffuse cortical a-β deposits were observed in one-third of the subjects (aged 35–62 years) as early as 2 h after injury [18]. Cortical a-β aggregates consist of amyloid peptides and hyperphosphorylated tau inclusions [19]. The currently prevailing hypothesis regarding AD pathogenesis is ‘amyloid cascade hypothesis’ stating: a-β peptides aggregate to form toxic oligomers and plaques which in turn lead to a cascade of neuropathological events, including neuroinflammation, oxidative stress, tau hyperphosphorylation and formation of neurofibrillary tangles, resulting ultimately in widespread neurodegeneration and dementia [20]. This hypothesis has been repeatedly revised, with the most prominent revision by Selkoe et al. in 2016, concluding that a-β dyshomeostasis had emerged as the most extensively validated and compelling therapeutic target from the whole cascade [21]. In a recent study using the (11)C-Pittsburgh compound B ((11)C-PiB)-PET in subjects 11 months to 17 years after TBI, increased burden of a-β was observed, mainly in the striatum and cerebellum [19]. Increased deposition of hyperphosphorylated tau is also a hallmark pathological marker of CTE as well as AD [11].
Considering that the secondary injury cascade after TBI plays a pivotal role in cognitive deficits in the short-term and long-term measure, correctly timed and targeted treatment may stop subsequent neuronal injury, leading to improvement of clinical symptoms of the patient. However, so far, no treatments for TBI have been successfully translated from testing on animal models through to the clinical phase. Past studies have often lacked the full pre-clinical evaluation with appropriate dose/response relationships and pharmacokinetic dynamics, extensively discussed elsewhere [22]. Still, there is a huge demand for treatment modalities that would improve patient outcome, both from a short-term and long-term perspective.
Oxidative stress and TBI
Oxidative stress and downstream oxidative and nitrative protein/lipid modifications that ensue are critical steps in the progression of neurodegenerative diseases and conditions [23]. Downstream changes may include misfolding and aggregation of proteins resulting in the subsequent accumulation of abnormal deposits, a core morphological feature of numerous neurodegenerative diseases, such as AD, PD or ALS [24]. More specifically, oxidative stress may induce (i) aberrant protein interactions, (ii) glial inflammation and (iii) may induce programmed cell death. Brain neurons are to some extent able to cope with increased oxidative stress: however, due to a phenomenon called selective neuronal vulnerability, some populations of neurons are more prone to the oxidative damage than others [25]. Yet, it is largely a mystery why some of the neuronal populations express such selective vulnerability; the genetic basis of which can explain only part of the observed variability [25]. A hallmark of oxidative damage in neurodegenerative diseases is lipid modification. Peroxidation, as a result of an attack by radicals on the double bond of unsaturated fatty acids located within neuronal membranes, generates highly reactive lipid peroxy radicals. ROS can initiate a cascade of reactions resulting in further modifications of polyunsaturated fatty acids (PUFAs) also in the membranes of nervous tissue cells. Such cascade reactions lead to the formation of breakdown products, e.g. 4-hydroxy-2,3-nonenal (HNE), malondialdehyde (MDA), acrolein [26] and F2-isoprostanes [27]. Given that the common understanding of oxidative stress as a key player in excitotoxicity and reperfusion injury, it is highly likely that oxidative stress plays a central role in secondary neuronal injury following TBI [28].
The increased sensitivity of neurons to oxidative stress is unsurprising, as (i) neurons are largely dependent on energy from aerobic metabolic pathways; (ii) nervous tissue has a larger content of lipid substances compared to other tissues, especially rich in polyunsaturated fatty acids; (iii) nervous tissue is generally exposed to high concentration of oxygen, receiving more than 20% of uptaken oxygen; (iv) nervous tissue has higher intracellular content of metals that can act as catalysts in the formation of reactive species and (v) nervous tissues contain a relatively smaller amount of antioxidants than other tissues [23]. In contrast to several proposed findings, evidence of elevated oxidative stress does not necessarily prove that it is involved in the neurodegeneration indicative of AD, PD, etc. It must be further elucidated as to whether the oxidative stress is a primary cause or merely a downstream consequence of the neurodegenerative process, as evidence is available for both hypotheses [24].
Vitamin E
Vitamin E is a potent antioxidant, soluble in lipids, preventing the propagation of free radicals and subsequent changes in membranes as well as plasmatic lipoproteins. The actions of vitamin E are not limited to antioxidant properties, as other non-antioxidant effects on signal transduction have been described, too. For a long time, most research on vitamin E has primarily focused on α-tocopherol (α-T), as α-T is the predominant form of vitamin E in tissues, and low intake of this form was associated with the clinical phenotype of vitamin E deficiency. Currently, more attention is being paid to other isoforms, especially γ-tocopherol (γ-T) that is naturally present in numerous dietary resources. However, only the naturally occurring RRR-α-tocopherol is considered to be the most physiologically active vitamer, with blood α-T concentrations being maintained by the preferential binding of α-T (compared to other tocopherols or tocotrienols) to the α-tocopherol transfer protein (α-TTP) [29].
Natural forms of vitamin E occur predominantly in oily plants. Although α-T exists in low concentrations in some fruits and vegetables, plant seeds, nuts and oils are the most abundant resources of α-T and γ-T. It has been described that while almonds, sunflower seeds and peanuts contain high amounts of α-T; pistachios, pecans, walnuts and palm oil contain mainly γ-T [30]. By virtue, α-T and γ-T are found in many common food oils, e.g. sunflower oil, peanut oil and soybean oil. As with other vitamins, the actual concentration of vitamin E obtained from the diet is dependent on food processing procedures (manufacture, cooking) as well as agricultural procedures (harvesting, growing) [31].
Vitamin E is primarily stored in adipose tissue (approximately 90% of the total body content of vitamin E) and symptoms of vitamin E deficiency are generally rare [32]. Severe vitamin E deficiency in humans is also relatively rare, occurring mostly as a result of rare mutations in the α-tocopherol transfer protein (α-TTP) gene, subsequently causing the AVED phenotype (OMIM #277460, ataxia with vitamin E deficiency). α-TTP is necessary to maintain normal plasmatic concentrations of α-T, while the absence of functional α-TTP leads to a profound drop in α-T levels [14]. Ulatowski and Manor [33] categorized metabolic deficiency of vitamin E into two groups—(i) primary deficiency as the result of inherited defects in proteins participating in vitamin E absorption/metabolization/elimination and (ii) secondary deficiency due to non-primary defects, mainly due to aberrant lipid metabolism, e.g. inherited defects in microsomal triglyceride transfer protein (MTTP), apoB mutations, cystic fibrosis or cholestasis [34].
The role of vitamin E in oxidative stress
Due to its physical-chemical properties, mainly the presence of a chromanol ring, α-T is an integral part of the antioxidant defence system, acting both as a lipid-soluble non-specific chain-breaking antioxidant and able to cooperate with a network of endogenous and exogenous, mainly dietary, antioxidants (Fig. 1). After reaction with the peroxyl radical (ROO-), the highly stable nascent α-tocopheroxyl radical is formed, and thus, the oxidative chain stops [35]. The vitamin is a typical peroxyl radical scavenger, and its main function is to protect PUFAs within membrane phospholipids, especially arachidonic acid (ARA, 20:4 ω-6) and docosahexaenoic acid (DHA, 22:6 ω-3). It has to be emphasized that these bioactive membrane lipids are important signalling molecules and that their modification due to oxidation is a key cellular event starting numerous signalling pathways within the cell itself [36].

Antioxidant defence system acting as a lipid-soluble non-specific chain-breaking antioxidant and cooperating with a network of endogenous and exogenous substances
Quite recently, vitamin E has started to attract more attention due to its antioxidant activity on reactive nitrogen species (RNS), including nitric oxide (NO), nitrogen dioxide (NO2) as well as peroxynitrite (ONOOO-) [37]. Unlike α-T, γ-T generally traps RNS including nitrogen dioxide and peroxynitrite to form a stable product, 5-NO2-γ-tocopherol (NGT) [38]. Later on, it was demonstrated that α-T supplementation in smokers attenuates γ-T scavenging of RNS by decreasing the availability of γ-T [38]. More research is definitely warranted to better understand the interplay between various isoforms of tocopherol in scavenging of ROS and RNS.
Vitamin E and non-TBI dementia
Even though the phenotype of the AD and non-TBI dementias differs from TBI-associated dementia, considerable evidence comes from both AD and non-AD dementia studies. Several animal studies also highlight the importance of vitamin E in AD. Mice exhibiting transgenic mouse model for Alzheimer’s disease (Papaw model), crossed with α-tocopherol transfer protein knock-out mice, displayed a significant increase in lipid peroxidation in specific brain regions [39]. The crossed double-mutant (type(−/−)Papaw) mice expressed heavy deposition of a-β plaques in the brain, this, however, could be substantially ameliorated with α-T supplementation. In another study on mice, axonal injury in the hippocampal CA1 region was observed in wild-type ageing mice, as well as vitamin-E deficient mice [40]. Since the hippocampus is associated to memory and cognition, this observation is consistent with the findings of hippocampal changes seen in the AD. Another study by Media et al. reports the reversal of detrimentally affected temporal discrimination, attributable to aggregated amyloid beta [41], after bilateral intrahippocampal injections by daily treatment with α-T [41].
However, it is impossible to derive a simple conclusion on the profits of vitamin E supplementation in AD. In a recent study by Arlt et al., the biochemical and clinical effect of vitamin E supplementation was investigated using a 1-year open clinical trial design [42], however, with negative conclusions. Vina et al. describe the ‘vitamin E paradox’, which says that in some patients, vitamin E could actually worsen the cognitive functions; whereas in others, vitamin E treatment partially prevents memory loss, typical for progression of the disease [43]. Observatory study from 2004 reports reduced levels of α-T in blood in women with the late-onset AD without vascular impairment [44].
In a post-mortem study on 230 subjects from the Religious Orders Study, ventricular cerebrospinal fluid (vCSF) obtained at autopsy was analyzed for α-T and γ-T in relation to brain tissue neuropathological diagnoses (NIA-Reagan criteria); density of neuritic plaques and Braak stage (neurofibrillary tangle stage); along with cognitive function (impairment/decline) before death [45]. In this study, higher α-T in vCSF was associated with better performance on tests of perceptual speed before death. Moreover, α-T levels were associated with less neuropathology typical for Alzheimer’s disease, specifically neuritic plaques.
There are scarce reports on an association of α-T with the non-AD type of dementia, most being published on vascular dementia (VaD). A systematic review of literature reports a protective effect of vitamin E in VaD [46]. In an older study from 2002 comparing circulating α-T in VaD with AD and control subjects, α-T was significantly lower in patients with VaD in comparison to patients with AD and controls. The α-T values for respective groups being 9.9, 12.6 and 12.6 ng/ml [47]. In a study on mice, supplementation with α-T (2 mM), α-tocotrienol (0.2 and 2 mM) and γ-T (0.2 and 2 mM) led to substantial decrease in size of cerebral infarcts 24 h after the occlusion of arteria cerebri media (MCA), while γ-tocotrienol, δT and δ-tocotrienol did not display any effects on the size or morphology of cerebral infarctions.
Possible mechanisms of vitamin E prophylactic effects in TBI
As previously mentioned, acute TBI is a complex disease characterized by two phases—primary and secondary. The primary phase of TBI is caused by direct injury to cells/tissues at the time of the initial event, which thus sparks an extensive series of biochemical pathways, leading to the subsequent development of secondary brain injury which basically consists of an ischaemic, inflammatory and cytotoxic component [48]. After the initial primary phase of injury, a complex cascade of cellular inflammation follows, leading to the progression of secondary damage. This inflammatory phase is typically systemic, i.e. not limited to the CNS, and involves a number of cytokines, chemokines, catecholamines and neurokinins, produced in the brain and elsewhere [39].
The core component of the secondary TBI phase is an acute depletion of energy which features an excessive influx of calcium, indicative of severe mitochondrial dysfunction leading to the inevitable activation of proteases. High intracellular ionic calcium is in turn associated with disruption of the cytoskeleton and the triggering of apoptotic pathways [49]. The massive energy crisis which follows TBI is generally associated with compromised synaptic plasticity, resulting in cognitive dysfunction [50]. In TBI animals, a decrease in the expression of AMP-activated protein kinase (AMPK), a major cellular energy sensor, is associated with the reduction of Sir2 and its downstream targets in proportion to the increased oxidative stress. It has been suggested by Aiguo et al. [51] that the levels of superoxide dismutase (SOD) and Sir2 (possibly involved in epigenetic gene silencing) are reduced in proportion to an increase in levels of protein oxidation after TBI. The same team suggested that dietary supplementation of vitamin E is capable of protecting against massive TBI by upregulating Sir2 and SOD [51]. While Sir2 contributes to elevation in antioxidant systems such as SOD, thus playing the crucial role in stress resistance and synaptic plasticity, the SOD protein is an important mitochondrial enzyme that acts as a scavenger of excessive ROS. As both these proteins are intercorrelated, an increase in Sir2 could well be driving the upregulation of SOD, conferring higher resistance to stress after TBI. The upregulation of SOD1, SOD2, as well as Sir2 has been associated with the anti-ageing effects of phloridzin in the yeast, Saccharomyces cerevisiae, and implicated in potentially anti-ageing effects of several other compounds [51]. However, more research into the role of Sir1/2 as well as SOD1/SOD2 in oxidative stress is warranted to fully elucidate their role.
Another possible mechanism for reducing the risk of the development of severe secondary TBI effects, including dementia, is the relationship between brain-derived neurotrophic factor (BDNF) and vitamin E supplementation. It is widely accepted that oxidative stress may disrupt cognitive functions and neuroelectricity by compromising the BNDF pathway. The BDNF system is a potent mechanism for the maintenance of functional and viable populations of neurons since BDNF promotes excitability as well as synaptic function. In the experiment conducted by Aigou et al. [51], who used dietary supplementation of vitamin E to protect against TBI-associated oxidative stress, supplementation was found to significantly normalize oxidative stress levels as well as limit neurological damage. Apart from the effect on Sir2/SOD that was discussed above, another possible explanation could be that vitamin E affected BDNF expression by influencing distinct subpopulations of neurons, thus promoting their survival/function.
Studies of vitamin E supplementation
So far, published reports on effects of vitamin E supplementation in the protection against consequences of TBI are scarce, contradictory and most are based on animal experiments. The question is what the right dose for the supplementation should be, which is a particularly complicated question in the animal models of TBI. In the study by Aigui et al. [51], rats were given 500 IU/kg of dietary of vitamin E for 4 weeks with subsequent TBI infliction. In another study, it has been reported that pre-conditioning of the CNS tissue with vitamin E significantly decreased brain lipid peroxidation and learning deficits in mouse model of ageing [52]. The study concludes that the exacerbation of brain oxidative stress following TBI plays a mechanistic role in accelerating amyloid-beta accumulation and associated behavioural impairments in the Tg2576 mice. In a recent study on α-tocopherol transfer protein-deficient mice, at 48 h-post-stroke, the S100B and RAGE expression were increased in stroke-affected cortices of mice with elevated brain α-T levels, and these increases were accompanied by pronounced microglial activation and neuroinflammatory signalling, typical hallmarks of brain damage [53]. Hence, it seems that high-dose α-T supplementation can have undesired consequences. In light of this, it seems logical that the exacerbation of microglial activation by excessive α-T depends on its unique cell signalling and independent of antioxidant functions of α-T [53].
Another study in albino rats subjected to TBI showed that supplementation with α-T statistically significantly reduced malondialdehyde in rats treated with 45 and 60 mg/kg body weight of ascorbic acid, α-tocopherol or a combination of the two vitamins for 2 weeks pre- and post-injury, compared with the group not subjected to TBI [54]. Another study by Yang et al. on the rat model of TBI showed α-T reduced Nogo-A and NgR expressions in brain tissue after TBI, also reporting that α-T promoted nerve regeneration when administered daily at the dose of 600 mg/kg [55]. Importantly, even though these studies present some evidence that α-T may play a beneficial role in TBI recovery, it is unclear whether these effects are associated with protection against long-term detrimental effects of TBI, such as early onset dementia, in humans.
Prior to discussing the issue of vitamin E supplementation/administration in humans with TBI, it is first essential to address the general issues revolving around supplementation in humans. Vitamin E supplementation designed to prevent TBI-associated dementia is generally based on estimations of adequate intake, which is a highly complex problem in the case of vitamin E. The determination of how much vitamin E is adequate depends not only on a specific assessment of its function, but also on defining a biomarker indicative of inadequacy which changes with nutrient intake [56]. The identification of such a biomarker is highly complicated and makes any conclusions associated with vitamin E supplementation/deficiency rather difficult. The estimation of a Recommended Dietary Allowance (RDA) by the Institute of Medicine (IOM) from 2000 was based on the presumption that increased peroxide-induced erythrocyte hemolysis corresponds to an increase in erythrocyte fragility in vitamin E-deficient individuals and that supplementation with α-T leads to the reduction of symptoms in affected individuals [40]. In particular, IOM used and adapted data from an old study by Horwitt et al. [57], concluding that the plasma α-T concentration of 12 μmol/L was associated with an observation of in vitro hydrogen peroxide-induced haemolysis below 12% (generally considered normal). Moreover, in a report from 2000, IOM concluded that no clear evidence for assuming different requirements of different population groups is available [56].
Vitamin E is generally present in numerous dietary sources, often along with fats, and is often ingested along with PUFAs which are one of the primary targets of α-T antioxidant action. In a recent scientific opinion by EFSA, however, an expert panel stated that—based on available data—no conclusions can be derived with respect to the relationship between PUFA intake and α-T intake/requirement, neither on their own or in combination to derive the requirement for α-T in adults. It is of interest that plant derivatives rich in γ-T often also contain considerable levels of PUFAs, especially n-6 PUFAs, while plant oils rich in α-T contain rather monounsaturated fatty acids (MUFAs). This finding rather compromises conclusions derived from observational studies of vitamin E intake as vitamin E is very often present in various proportions of MUFAs/PUFAs in a range dietary resources, making it difficult to untangle possible synergism/antagonism between these fatty compounds. Based on an EFSA panel recommendation regarding vitamin E supplementation from 2015, recommended adequate intake (AI) values for vitamin E for the European population are as follows: 13 mg/day for adult males and 11 mg/day for adult females. For children aged 1 to 3 years, AI for α-T is 6 mg/day for both sexes. For children aged 3 to 10 years, AI for α-T is estimated as 9 mg/day, also for both sexes. For teenagers and adolescents aged 10 to 18 years, AI for α-T is estimated as 13 mg/day for males and 11 mg/day for females. For infants aged 7 to 11 months, an AI value for α-T of 5 mg/day is derived using extrapolation of estimated α-T l intake for exclusively breast-fed infants aged 0–6 months.
Only a limited number of studies with the appropriate randomized blinded clinical design focus on the effects of vitamin E dietary supplementation in TBI in humans. A single major trial was conducted in 2011 by Razmkon et al., who investigated the effects of vitamin E administration in the case of severe head injury [58]. In this study, a total of 100 study subjects were randomized into four groups subject to the following protocols: group A, low-dose vitamin C (500 mg/d IV) for 7 days; group B, high-dose vitamin C (10 g IV on the first (admission) day and repeated on the fourth day, followed up by vitamin C 4 g/d IV for the remaining 3 days); group C, vitamin E (400 IU/d IM) for 7 days; and group D, placebo. In this study, vitamin E was shown to affect mortality as well as improved functional outcomes at discharge. This effect seems to be of a rather short-term nature, as it diminished during the 6-month follow-up period. From this point of view, the effect of vitamin E on the prevention of dementia development on a long-term scale remains highly enigmatic.
Novel drug therapy based on vitamin E
Vitamin E-based lazaroids
While studies incorporating the use of vitamin E in TBI as a prevention of TBI-associated comorbidities are very scarce, there are several studies associating the positive effects of administration of lazaroids, partially derived from the chromanol ring of α-T, in models based on post-ischaemic reperfusion injury and TBI [59,60,61,62]. Lazaroids are 21-aminosteroids which act as potent inhibitors of iron-dependent lipid peroxidation in various tissues, originally developed and indicated for the treatment of CNS conditions and ischaemia [63].
U-83836E in TBI
U-83836E is a second-generation synthetic dual mechanism lazaroid, characterized by the chemical combination of bis-pyrrolidino-pyrimidine, manifesting functionality from the lipid peroxidation inhibitor tirilazad, with the chromanol ring portion of α-T bonded to various amine groups [64]. This 2-methylaminochroman compound with its bis-pyrrolidino-pyrimidine moiety of tirilazad is capable of catalytically scavenging LOO• radical, while the electron-donating chromanol, after donating the phenolic electron to a LOO•, can be reduced by endogenous reducing agents such as glutathione or ascorbate [65]. This free radical scavenger has potent neuroprotective effects against ischaemic injury and diabetic neuropathy [62, 66]. Eminently, at higher doses, U-83836E is highly cytotoxic, which can be demonstrated, for example, by its ability to reduce the viability of rat brain endothelial cells (RBE4) with an IC50 value of 0.032 mM [67]. U-83636E is highly lipophilic with a high affinity for membrane phospholipids that are mostly exposed to lipid peroxidation, making it a good candidate for TBI treatment.
Several studies investigated the potential of U-83636E for inhibiting lipoperoxidation in TBI. In 2010, Mustafa et al. demonstrated that U-83836E treatment can promote the inhibition of post-traumatic lipoperoxidation in cerebral cortical tissue or mitochondria while preserving aerobic respiratory function and Ca++-buffering capacity [65]. Shortly afterwards, the same research group suggested that U-83836E can attenuate calpain-mediated cytoskeletal damage in the same animal model, at least in part by means of mitochondrial functional protection with U-83836E-repeated dosing within the therapeutic 12-h window [68]. In this regard, U-83836E is able to inhibit an early event in a series of linked secondary injury pathways, thereby providing neuroprotection at multiple biochemical levels. Mustafa et al. [68] further suggest that in order for U-83836E to be effective when treatment is delayed beyond 12 h after the event, it is necessary that intensive (i.e. repeated) dosing be applied to stop the likely intense lipid peroxidation that has almost certainly developed at approximately 12-h post-injury.
Generally speaking, U-83836E certainly represents a very potent inhibitor of lipid peroxidation in vivo. Its cytotoxicity, however, is currently limiting its possible clinical use; therefore, research into its role in the prevention of TBI-associated pathological events is imperative.
Conclusion
Vitamin E and its derived molecules present an elegant candidate for a protective substance against long-term comorbidity associated with TBI. Never, more research into the role of vitamin E and associated molecules in TBI is necessary.
References
Cassidy JD, Carroll LJ, Peloso PM, Borg J, von Holst H, Holm L, Kraus J, Coronado V (2004) Incidence, risk factors and prevention of mild traumatic brain injury: results of the WHO Collaborating Centre Task Force on Mild Traumatic Brain Injury. J Rehabil Med 36:28–60
Li W, Risacher SL, McAllister TW, Saykin AJ (2016) Traumatic brain injury and age at onset of cognitive impairment in older adults. J Neurol 263:1280–1285. https://doi.org/10.1007/s00415-016-8093-4
LoBue C, Cullum CM, Didehbani N et al (2017) Neurodegenerative dementias after traumatic brain injury. J Neuropsychiatry Clin Neurosci. https://doi.org/10.1176/appi.neuropsych.17070145
Wood RL (2017) Accelerated cognitive aging following severe traumatic brain injury: a review. Brain Inj 31:1270–1278. https://doi.org/10.1080/02699052.2017.1332387
Murray GD, Teasdale GM, Braakman R, Cohadon F, Dearden M, Iannotti F, Karimi A, Lapierre F, Maas A, Ohman J, Persson L, Servadei F, Stocchetti N, Trojanowski T, Unterberg A (1999) The European Brain Injury Consortium survey of head injuries. Acta Neurochir 141:223–236
Tagliaferri F, Compagnone C, Korsic M, Servadei F, Kraus J (2006) A systematic review of brain injury epidemiology in Europe. Acta Neurochir 148:255–268; discussion 268. https://doi.org/10.1007/s00701-005-0651-y
Peeters W, van den Brande R, Polinder S, Brazinova A, Steyerberg EW, Lingsma HF, Maas AIR (2015) Epidemiology of traumatic brain injury in Europe. Acta Neurochir 157:1683–1696. https://doi.org/10.1007/s00701-015-2512-7
Andriessen TMJC, Jacobs B, Vos PE (2010) Clinical characteristics and pathophysiological mechanisms of focal and diffuse traumatic brain injury. J Cell Mol Med 14:2381–2392. https://doi.org/10.1111/j.1582-4934.2010.01164.x
AC M, Daneshvar DH (2015) The neuropathology of traumatic brain injury. Handb Clin Neurol 127:45–66. https://doi.org/10.1016/B978-0-444-52892-6.00004-0
Bullock R, Chesnut RM, Clifton G et al (1996) Guidelines for the management of severe head injury. Brain Trauma Foundation. Eur J Emerg Med Off J Eur Soc Emerg Med 3:109–127
Corrigan F, Arulsamy A, Teng J, Collins-Praino LE (2017) Pumping the brakes: neurotrophic factors for the prevention of cognitive impairment and dementia after traumatic brain injury. J Neurotrauma 34:971–986. https://doi.org/10.1089/neu.2016.4589
Loane DJ, Faden AI (2010) Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci 31:596–604. https://doi.org/10.1016/j.tips.2010.09.005
Rabinowitz AR, Levin HS (2014) Cognitive sequelae of traumatic brain injury. Psychiatr Clin N Am 37:1–11. https://doi.org/10.1016/j.psc.2013.11.004
Brown AW, Moessner AM, Mandrekar J, Diehl NN, Leibson CL, Malec JF (2011) A survey of very-long-term outcomes after traumatic brain injury among members of a population-based incident cohort. J Neurotrauma 28:167–176. https://doi.org/10.1089/neu.2010.1400
Cantu RC (2001) Posttraumatic retrograde and anterograde amnesia: pathophysiology and implications in grading and safe return to play. J Athl Train 36:244–248
Smith DH, Johnson VE, Stewart W (2013) Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol 9:211–221. https://doi.org/10.1038/nrneurol.2013.29
Gardner RC, Burke JF, Nettiksimmons J, Kaup A, Barnes DE, Yaffe K (2014) Dementia risk after traumatic brain injury vs nonbrain trauma: the role of age and severity. JAMA Neurol 71:1490–1497. https://doi.org/10.1001/jamaneurol.2014.2668
Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VMY, Clark RS, Marion DW, Wisniewski SR, DeKosky ST (2004) Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol 190:192–203. https://doi.org/10.1016/j.expneurol.2004.06.011
Scott G, Ramlackhansingh AF, Edison P, Hellyer P, Cole J, Veronese M, Leech R, Greenwood RJ, Turkheimer FE, Gentleman SM, Heckemann RA, Matthews PM, Brooks DJ, Sharp DJ (2016) Amyloid pathology and axonal injury after brain trauma. Neurology 86:821–828. https://doi.org/10.1212/WNL.0000000000002413
Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256:184–185
Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8:595–608. https://doi.org/10.15252/emmm.201606210
Chakraborty S, Skolnick B, Narayan RK (2016) Neuroprotection trials in traumatic brain injury. Curr Neurol Neurosci Rep 16:29. https://doi.org/10.1007/s11910-016-0625-x
Chen X, Guo C, Kong J (2012) Oxidative stress in neurodegenerative diseases. Neural Regen Res 7:376–385. https://doi.org/10.3969/j.issn.1673-5374.2012.05.009
Andersen JK (2004) Oxidative stress in neurodegeneration: cause or consequence? Nat Med 10(Suppl):S18–S25. https://doi.org/10.1038/nrn1434
Hardy J (2016) Catastrophic cliffs: a partial suggestion for selective vulnerability in neurodegenerative diseases. Biochem Soc Trans 44:659–661. https://doi.org/10.1042/BST20150287
Singh M, Dang TN, Arseneault M, Ramassamy C (2010) Role of by-products of lipid oxidation in Alzheimer’s disease brain: a focus on acrolein. J Alzheimers Dis 21:741–756. https://doi.org/10.3233/JAD-2010-100405
Galasko DR, Peskind E, Clark CM, Quinn JF, Ringman JM, Jicha GA, Cotman C, Cottrell B, Montine TJ, Thomas RG, Aisen P, Alzheimer’s Disease Cooperative Study (2012) Antioxidants for Alzheimer’s disease: a randomized clinical trial with cerebrospinal fluid biomarker measures. Arch Neurol 69:836–841. https://doi.org/10.1001/archneurol.2012.85
Cruz-Haces M, Tang J, Acosta G, Fernandez J, Shi R (2017) Pathological correlations between traumatic brain injury and chronic neurodegenerative diseases. Transl Neurodegener 6:20. https://doi.org/10.1186/s40035-017-0088-2
Hosomi A, Arita M, Sato Y, Kiyose C, Ueda T, Igarashi O, Arai H, Inoue K (1997) Affinity for alpha-tocopherol transfer protein as a determinant of the biological activities of vitamin E analogs. FEBS Lett 409:105–108
McLaughlin PJ, Weihrauch JL (1979) Vitamin E content of foods. J Am Diet Assoc 75:647–665
Dutta A, Dutta SK (2003) Vitamin E and its role in the prevention of atherosclerosis and carcinogenesis: a review. J Am Coll Nutr 22:258–268
Schmölz L, Birringer M, Lorkowski S, Wallert M (2016) Complexity of vitamin E metabolism. World J Biol Chem 7:14–43. https://doi.org/10.4331/wjbc.v7.i1.14
Ulatowski L, Manor D (2013) Vitamin E trafficking in neurologic health and disease. Annu Rev Nutr 33:87–103. https://doi.org/10.1146/annurev-nutr-071812-161252
Tanyel MC, Mancano LD (1997) Neurologic findings in vitamin E deficiency. Am Fam Physician 55:197–201
Burton GW, Joyce A, Ingold KU (1982) First proof that vitamin E is major lipid-soluble, chain-breaking antioxidant in human blood plasma. Lancet 2:327
Traber MG, Atkinson J (2007) Vitamin E, antioxidant and nothing more. Free Radic Biol Med 43:4–15. https://doi.org/10.1016/j.freeradbiomed.2007.03.024
Christen S, Jiang Q, Shigenaga MK, Ames BN (2002) Analysis of plasma tocopherols alpha, gamma, and 5-nitro-gamma in rats with inflammation by HPLC coulometric detection. J Lipid Res 43:1978–1985
Christen S, Woodall AA, Shigenaga MK, Southwell-Keely PT, Duncan MW, Ames BN (1997) γ-Tocopherol traps mutagenic electrophiles such as NOx and complements α-tocopherol: physiological implications. Proc Natl Acad Sci U S A 94:3217–3222
Nishida Y, Yokota T, Takahashi T, Uchihara T, Jishage KI, Mizusawa H (2006) Deletion of vitamin E enhances phenotype of Alzheimer disease model mouse. Biochem Biophys Res Commun 350:530–536. https://doi.org/10.1016/j.bbrc.2006.09.083
Fukui K, Kawakami H, Honjo T et al (2012) Vitamin E deficiency induces axonal degeneration in mouse hippocampal neurons. J Nutr Sci Vitaminol (Tokyo) 58:377–383
McDaid DG, Kim E-M, Reid RE et al (2005) Parenteral antioxidant treatment preserves temporal discrimination following intrahippocampal aggregated Abeta(1-42) injections. Behav Pharmacol 16:237–242
Arlt S, Müller-Thomsen T, Beisiegel U, Kontush A (2012) Effect of one-year vitamin C- and E-supplementation on cerebrospinal fluid oxidation parameters and clinical course in Alzheimer’s disease. Neurochem Res 37:2706–2714. https://doi.org/10.1007/s11064-012-0860-8
Viña J, Lloret A, Giraldo E et al (2011) Antioxidant pathways in Alzheimer’s disease: possibilities of intervention. Curr Pharm Des 17:3861–3864
Glasø M, Nordbø G, Diep L, Bøhmer T (2004) Reduced concentrations of several vitamins in normal weight patients with late-onset dementia of the Alzheimer type without vascular disease. J Nutr Health Aging 8:407–413
Hensley K, Barnes LL, Christov A, Tangney C, Honer WG, Schneider JA, Bennett DA, Morris MC (2011) Analysis of postmortem ventricular cerebrospinal fluid from patients with and without dementia indicates association of vitamin E with neuritic plaques and specific measures of cognitive performance. J Alzheimers Dis 24:767–774. https://doi.org/10.3233/JAD-2011-101995
Perez L, Heim L, Sherzai A et al (2012) Nutrition and vascular dementia. J Nutr Health Aging 16:319–324
Ryglewicz D, Rodo M, Kunicki PK et al (2002) Plasma antioxidant activity and vascular dementia. J Neurol Sci 203–204:195–197
Veenith T, Goon SS, Burnstein RM (2009) Molecular mechanisms of traumatic brain injury: the missing link in management. World J Emerg Surg 4:7. https://doi.org/10.1186/1749-7922-4-7
Bondi CO, Semple BD, Noble-Haeusslein LJ, Osier ND, Carlson SW, Dixon CE, Giza CC, Kline AE (2015) Found in translation: understanding the biology and behavior of experimental traumatic brain injury. Neurosci Biobehav Rev 58:123–146. https://doi.org/10.1016/j.neubiorev.2014.12.004
Agrawal R, Tyagi E, Vergnes L, Reue K, Gomez-Pinilla F (2014) Coupling energy homeostasis with a mechanism to support plasticity in brain trauma. Biochim Biophys Acta (BBA) - Mol Basis Dis 1842:535–546. https://doi.org/10.1016/j.bbadis.2013.12.004
Wu A, Ying Z, Gomez-Pinilla F (2010) Vitamin E protects against oxidative damage and learning disability after mild traumatic brain injury in rats. Neurorehabil Neural Repair 24:290–298. https://doi.org/10.1177/1545968309348318
Conte V, Uryu K, Fujimoto S, Yao Y, Rokach J, Longhi L, Trojanowski JQ, Lee VMY, McIntosh TK, Pratico D (2004) Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. J Neurochem 90:758–764. https://doi.org/10.1111/j.1471-4159.2004.02560.x
Khanna S, Heigel M, Weist J, Gnyawali S, Teplitsky S, Roy S, Sen CK, Rink C (2015) Excessive α-tocopherol exacerbates microglial activation and brain injury caused by acute ischemic stroke. FASEB J 29:828–836. https://doi.org/10.1096/fj.14-263723
Ishaq GM, Saidu Y, Bilbis LS et al (2013) Effects of α-tocopherol and ascorbic acid in the severity and management of traumatic brain injury in albino rats. J Neurosci Rural Pract 4:292–297. https://doi.org/10.4103/0976-3147.118784
Yang J, Han Y, Ye W, Liu F, Zhuang K, Wu G (2013) Alpha tocopherol treatment reduces the expression of Nogo-A and NgR in rat brain after traumatic brain injury. J Surg Res 182:e69–e77. https://doi.org/10.1016/j.jss.2012.11.010
Institute of Medicine (US) Panel on Dietary Antioxidants and Related Compounds (2000) Dietary reference intakes for vitamin C, vitamin E, selenium, and carotenoids. National Academies Press (US), Washington
Horwitt MK, Century B, Zeman AA (1963) Erythrocyte survival time and reticulocyte levels after tocopherol depletion in man. Am J Clin Nutr 12:99–106
Razmkon A, Sadidi A, Sherafat-Kazemzadeh E, Mehrafshan A, Jamali M, Malekpour B, Saghafinia M (2011) Administration of vitamin C and vitamin E in severe head injury: a randomized double-blind controlled trial. Clin Neurosurg 58:133–137
Durmaz R, Ertilav K, Akyüz F, Kanbak G, Bildirici K, Tel E (2003) Lazaroid U-74389G attenuates edema in rat brain subjected to post-ischemic reperfusion injury. J Neurol Sci 215:87–93
Durmaz R, Kanbak G, Akyüz F, Isiksoy S, Yücel F, Inal M, Tel E (2003) Lazaroid attenuates edema by stabilizing ATPase in the traumatized rat brain. Can J Neurol Sci J Can Sci Neurol 30:143–149
Tseng MT, Chan SA, Reid K, Lyer V (1997) Post-ischemic treatment with a lazaroid (U74389G) prevents transient global ischemic damage in rat hippocampus. Neurol Res 19:431–434
Lai L-N, Zhang X-J, Zhang X-Y et al (2016) Lazaroid U83836E protects the heart against ischemia reperfusion injury via inhibition of oxidative stress and activation of PKC. Mol Med Rep 13:3993–4000. https://doi.org/10.3892/mmr.2016.5030
Braughler JM, Pregenzer JF (1989) The 21-aminosteroid inhibitors of lipid peroxidation: reactions with lipid peroxyl and phenoxy radicals. Free Radic Biol Med 7:125–130
Hall ED, Braughler JM, Yonkers PA, Smith SL, Linseman KL, Means ED, Scherch HM, von Voigtlander P, Lahti RA, Jacobsen EJ (1991) U-78517F: a potent inhibitor of lipid peroxidation with activity in experimental brain injury and ischemia. J Pharmacol Exp Ther 258:688–694
Mustafa AG, Singh IN, Wang J, Carrico KM, Hall ED (2010) Mitochondrial protection after traumatic brain injury by scavenging lipid peroxyl radicals. J Neurochem 114:271–280. https://doi.org/10.1111/j.1471-4159.2010.06749.x
Wang D, Qu Z, Yang L, Zhang Q, Liu ZH, Do T, Adelson DL, Wang ZY, Searle I, Zhu JK (2017) Transposable elements (TEs) contribute to stress-related long intergenic noncoding RNAs in plants. Plant J Cell Mol Biol 90:133–146. https://doi.org/10.1111/tpj.13481
Blasig IE, Mertsch K, Haseloff RF (2002) Nitronyl nitroxides, a novel group of protective agents against oxidative stress in endothelial cells forming the blood-brain barrier. Neuropharmacology 43:1006–1014
Mustafa AG, Wang JA, Carrico KM, Hall ED (2011) Phramacological inhibition of lipid peroxidation attenuates calpain-mediated cytoskeletal degradation after traumatic brain injury. J Neurochem 117:579–588. https://doi.org/10.1111/j.1471-4159.2011.07228.x
Acknowledgements
We would like to thank the CETOCOEN PLUS project (CZ.02.1.01/0.0/0.0/15_003/0000469). The RECETOX research infrastructure was supported by the Ministry of Education, Youth and Sports of the Czech Republic (LM2011028).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Highlights
• Vitamin E can be protective against traumatic brain injury (TBI)-associated dementia
• Animal models also support the hypothesis vitamin E has protective effects against TBI-dementia
• Effects of dietary supplementation of vitamin E on TBI-associated dementia are discussed
Rights and permissions
About this article

Cite this article
Dobrovolny, J., Smrcka, M. & Bienertova-Vasku, J. Therapeutic potential of vitamin E and its derivatives in traumatic brain injury-associated dementia. Neurol Sci 39, 989–998 (2018). https://doi.org/10.1007/s10072-018-3398-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-018-3398-y