
Abstract
The mutations of MYH7 (slow skeletal/β-cardiac myosin heavy chain) are commonly found in familial hypertrophic/dilated cardiomyopathy, and also can cause Laing early-onset distal myopathy (LDM), myosin storage myopathy (MSM), and congenital myopathy with fiber-type disproportion (CFTD). Here we report two cases whose diagnosis was hereditary myopathy according to clinical feature and muscle pathology analysis. High-throughput genomic sequencing (next generation sequencing) was performed to validate the diagnosis. Two MYH7 mutations, p.R1845W and p.E1687del, were identified. p.R1845W was found in a male patient showing weakness of both terminal lower legs without foot drop. Muscle pathology stainings characteristically showed the hyaline body in the intracytoplasmic location. The novel mutation p.E1687del was found in a family with seven patients. The proband showed foot drop, scoliosis, and winged scapula, while his mother only showed mild foot drop and winged scapula. Muscle pathology analysis showed congenital centronucleus myopathy. Both cases only showed muscular disorder and had no cardiomyopathy. This study, for the first time, reports the MYH7 mutations associated with centronucleus myopathy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Myosin 7 gene (MYH7), located in 14q11.2, encodes slow/beta-cardiac myosin heavy chain (MHC-β), a class II myosin expressed primarily in the heart, but also in skeletal muscles (particularly in type I fibers) [1]. Myosin is a major component of heart and skeletal muscle, and plays a key role in muscle contractility. More than 200 mutations of MYH7 have been identified; these are associated with a variety of clinical myosinopathies, including cardiac and skeletal myopathy [2].
Many studies have confirmed the phenotypic variability of MYH7 myopathy, which is believed to depend on the gene mutation site. Mutations in the globular head of the protein were shown to be linked with cardiomyopathies, including familial hypertrophic cardiomyopathy, dilated cardiomyopathy, and left ventricular non-compaction cardiomyopathy 4; mutations in the distal rod were shown to be linked to myosin storage myopathy (MSM), while those in middle or proximal rod cause Laing early-onset distal myopathy (LDM) or congenital myopathy with fiber-type disproportion (CFTD) with/without specific cardiac impairment [3,4,5,6]. Almost all mutations reported till date have a dominant effect, although cases of recessive inheritance as well as sporadic cases have also been documented [3, 7].
Patient and methods
Patients
We retrospectively reviewed clinical and pathological features of two patients with the diagnosis of hereditary myopathy. Clinical features and laboratory data of these patients were collected, including the general background genealogical data, MRI images, and results of histopathological examination of muscle biopsy. Written informed consent was obtained from all subjects. The study was approved by the Ethical Committee of the Third Hospital of Hebei Medical University and was conducted in accordance with the Declaration of Helsinki.
Methods
Muscle biopsy and histochemistry
Open muscle biopsy was performed in the biceps brachii muscles after obtaining the written informed consent from patients. The specimens were frozen and 7-μm-thick serial sections prepared. Staining was performed with hematoxylin and eosin (H&E) and modified Gomori trichrome (MGT). For nicotinamide adenine dinucleotide tetrazolium reductase (NADH-TR), succinic dehydrogenase (SDH), adenosine monophosphate deaminase, cytochrome c oxidase (CCO), acid phosphatase, and adenosine triphosphatase (ATPase), after incubation at pH 4.35, 4.6, and 10.0, the specimens were placed in fluids with specific enzyme substrate, respectively. Periodic acid–Schiff reaction was used to detect the inflammatory cells and phagocytic cells with increased enzymatic activity. Oil Red O and Sudan Black B were used to detect the degree of lipid deposition. Stained tissues were observed under a light microscope.
Next generation sequencing
DNA extraction and NGS library preparation
Genomic DNA (gDNA) was extracted from peripheral blood (derived from two patients and mother of patient 2) with a Mag-Bind Blood DNA Kit (CoWin Biotech). A minimum of 3 μg of DNA was used to generate the indexed Illumina sequencing libraries according to the manufacturer’s protocol. A library size of 300–400 bp, including adapter sequences, was finally selected.
Target gene capture and NGS
The genes related to myopathy (Table 1) were enriched using the GenCap capture probe (MyGenostics, Beijing, China). The biotinylated 100-mer oligo baits were designed to tile all the exon regions of the target genes. The capture experiment was conducted according to the manufacturer’s protocol. The enrichment paired-end libraries were sequenced on an Illumina HiSeq 2000 sequencer to obtain the reads of 100 bp.
Bioinformatics analysis
Clean sequencing reads were aligned to each human reference genome using the Burrows–Wheeler Alignment program (http://bio-bwa.sourceforge.net/bwa.shtml), and quality scores were recalibrated and realigned to the reference using GATK software. Duplicated reads were removed using Sequence Alignment/Map tools, and only uniquely mapped reads were used for variation detection. Single-nucleotide variants (SNVs) were detected and genotyped with the GATK UnifiedGenotyper. Small insertions/deletions (Indels) were detected with the GATK Indel Genotyper V2. Annotation of the variants, such as locations (exonic, intronic, and intergenic regions) and effects on protein coding (synonymous, missense, nonsense, and frameshift), was assessed with a bioinformatics tool developed in-house with RefSeq (hg19, from UCSC) and UCSC annotation (http://www.ncbi.nlm.nih.gov/refseq/). The SNVs/Indels were filtered if they showed > 5% frequency in several databases, including in the NCBI dbSNP138 (https://www.ncbi.nlm.nih.gov/snp), 1000 Genomes (http://www.internationalgenome.org/), and the 300 in-house Asia database (i.e., generated by NGS of DNA from 300 normal Chinese individuals, and provided by MyGenostics, Inc.). The probable pathogenic mutations were then predicted using SIFT (http://sift.jcvi.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and MutationTaster (http://mutationtaster.org/).
Sanger sequencing
Sanger sequencing was performed to validate the variants identified by NGS and to examine the available relatives of the patients. Amplified fragments were directly sequenced using BigDye Terminator Cycle Sequencing Kits on an automated genetic analyzer (ABI3130; Applied Biosystems, Foster City, California). Sequences were analyzed by Chromas software (http://technelysium.com.au/wp/chromas/) and compared with the reference sequences of MYH7.
Results
Clinical features
Patient 1 was a 46-year-old Chinese man who developed slow but progressive lower-extremity weakness with late onset. The weakness progressed over the past 6 years, affecting ambulation and elevation of his arms. The family history was normal. His son, aged 26, was clinically normal. Physical examination revealed high arches, waddling gait, weakness, and atrophy of pelvic and scapular muscles, especially both biceps femoris muscles and pseudohypertrophy of calf (Fig. 1). There were no signs of high-arched palate, tremors, sensory disturbances, contractures of joints, or scoliosis. His creatine kinase (CK) level was raised (529 U/L), and electromyography findings were consistent with chronic myopathy. MRI examination of thigh muscles showed fatty change of biceps femoris (BF), adductor longus (AL), adductor magnus (AM), sartorius (S), semimembranosus (SM), and semitendinosus (ST) muscles. The gracilis (G) showed mild changes, while the vastus intermedius (VI), vastus lateralis (VL), and vastus medialis (VM) muscles were relatively spared. The leg muscles that were predominantly affected included the medial gastrocnemius (GA), peroneal group (PG), and tibialis anterior (TA) muscles. The soleus (SO) was relatively spared (Fig. 2).

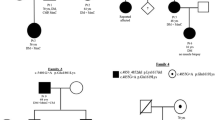
Clinical image of two patients and pedigree of the patient 2 family showing an autosomal dominant inheritance. A1: patient 1, A2: gastrocnemius hypertrophy, A3: posterior thigh muscle atrophy. B1: patient 2, standing position; B2: prone position

MRI images of muscles of two patients. Fatty change of biceps femoris (BF), adductor longus (AL), adductor magnus (AM), sartorius (S), semimembranosus (SM), and semitendinosus (ST) in thigh of patient 1, and medial gastrocnemius (GA), peroneal group (PG), and tibialis anterior (TA) muscles in calf of patient 1; and fatty change of both anterior tibial muscles in leg of patient 2
Patient 2: The boy belonged to a family with three generations affected by an autosomal dominant myopathy, which was characterized by onset at early childhood, foot drop, limb-girdle weakness, scoliosis, and slow progression (Fig. 1). Seven patients of the family had obvious muscular weakness and atrophy. They showed no dysarthria, dysphagia, or facial muscle involvement, and had no high-arched palate, tremors, sensory disturbances, contractures of joints, respiratory failures, or obvious cardiac failure. Their intelligence level was normal. As the patients’ residence was thousands of kilometers away, only III-9 and II-4 came to our hospital; other patients of the family were not assessed in this study. Examination of the boy (III-9) and his mother (II-4) revealed scoliosis. There was no weakness of both upper limbs, although he had left scapular wing. There was weakness in lower limb muscles, which was more marked in the tibialis anterior and posterior muscle group of thigh. Bilateral foot drop led to a special walking gait. There was no bilateral calf hypertrophy and no evidence of cardiomyopathy. His CK level was normal. Electromyography revealed obvious myopathy. MRI showed fatty change in both anterior tibial muscles. VI, VM, and BF were slightly involved; the predominantly affected leg muscle was TA, followed by medial gastrocnemius (Fig. 2).
Muscle pathology
Patient 1: Muscle biopsy showed numerous pale subsarcolemmal hyaline bodies, with positive myosin ATPase and negative oxidative enzymes staining present exclusively in type I muscle fibers (Fig. 3).

Histochemical staining of skeletal muscle. a Case 2, III-9, sections stained for NADH. b Case 2, III-9, sections stained for H&E. c Case 2, II-4, sections stained for H&E. d Case 1, sections stained for MGT. e Case 1, sections stained for H&E. f Case 1, sections stained for SDH. The arrows refer to pale subsarcolemmal hyaline bodies
Patient 2: Muscle biopsy showed non-specific myopathic changes with no signs of dystrophic or inflammatory changes; numerous rounded atrophic muscle fibers, isolated or grouped, were observed. Type I fiber predominance was noted and most atrophic fibers were type I. A tendency of fiber-type grouping was noted, without sarcoplasmic inclusions, cores, or minicores. In the NADH section, there were abnormalities in myofibril organization, such as eddy-like/Wipe pattern/myofibrillar netting disorders (Fig. 3).
Molecular genetic analyses
Patient 1: The sequencing analysis revealed a novel heterozygous point mutation at nucleotide 5533 with C to T transition in exon 37 of MYH7, resulting in an amino acid substitution p.R1845W. This mutation is a known pathogenic mutation that causes MSM [8]. This mutation lies at the tail of the myosin protein.
Patient 2: A novel 3-bp in-frame deletion, c.5059-5061del, was found in MYH7 gene on 14q11.2, which results in a p.E1687del amino acid deletion. This heterozygous mutation was further investigated by Sanger sequencing in his parents, and his mother was found to have the same mutation. Unfortunately, whether the deletion segregated with the disease could not be determined due to lack of samples from the other family members.
Discussion
Clinical phenotype
The MYH7 gene, encoding the slow/beta cardiac myosin heavy chain, is known as the cause of hypertrophic or dilated cardiomyopathy, LDM, and MSM. So far, more than 200 dominant mutations of MYH7 have been shown to be associated with cardiomyopathy; however, only a small number of these caused skeletal myopathy. Missense mutations in the globular head and tail of MYH7 are a frequent cause of familial hypertrophic cardiomyopathy [2]. In Asian people, although many MYH7 mutations have been reported to cause cardiomyopathy [9, 10], only a few familial cases of Korean and Chinese with LDM, and a sporadic case with MSM, were reported to carry a mutation in this gene. The Korean myopathy family with the p.A1439P MYH7 mutation was described to have prominent paraspinal and proximal muscle involvement [11]. The Chinese family with the p.K1617del MYH7 mutation presented as LDM with scoliosis and calf hypotrophy [12]. It was reported that a patient with the same mutation as that of patient 1 (p.R1845W) showed secondary changes of hypertension in electrocardiogram [8], with no associated evidence of cardiomyopathy, which is strikingly similar to that observed in our patient 1. These data suggest that the heart is slightly affected by this mutation. In our patient, the power of quadriceps femoris was normal, which is different from the known characteristics of late-onset MSM, as the main manifestation in the latter condition is quadriceps and iliopsoas involvement [8]. Our case may represent a new phenotype of MSM.
In our case 2, genetic analysis indicated autosomal dominant inheritance with childhood onset. Clinical manifestations were similar to congenital myopathy. The phenotype included weakness and atrophy of anterior compartment tibial muscles and toe extensor (dorsiflexor) muscles. Another prominent symptom was scoliosis, especially when the standing position was significantly heavier than the supine position, which indicated that the scoliosis was related to the erector spinae weakness and is suggestive of axial muscle involvement. The boy never complained of heart palpitations and chest distress, and the physical examination and echocardiogram of the heart were normal. In order to study the heart involvement with this mutation, we examined his mother, using electrocardiogram and echocardiogram, which confirmed the selective involvement of skeletal muscle and absence of any myocardial damage.
According to our cases and literature reports, the diversity of the phenotypes of MYH7-related myopathy is significant, indicating the high heterogeneity in clinical genetics [13, 14].
Pathology
Skeletal muscle biopsy in case 1 showed typical pathological features of hyaline body myopathy, which is consistent with previous reports [8]. In case 2, there were pathological changes of centronuclear myopathy (CNM), excessive variation in fiber size, with obvious group atrophy and type I fiber preponderance, and absence of sarcoplasmic inclusions. Core pathology was common in muscle biopsies of MYH7 mutation cases [5, 15, 16], and myofibrillar myopathy (MFM)-like changes were also present [17]. Many cases were diagnosed as CFTD before gene analysis [5, 18]. To date, there is no report on CMN caused by MYH7 mutation [19, 20]. It is postulated that these pathologies likely implicate a complex interaction between slow myosin and other sarcomeric proteins, and an impaired or insufficient degradation of myofibrillar proteins [17].
Overall, no typical abnormality in muscle pathology has emerged to help in a definitive diagnosis of LDM, in contrast to the allelic MSM that is defined by the presence of the subsarcolemmal hyaline bodies. Therefore, the pathological findings cannot be used to detect the target genes.
Gene
We found a novel non-frameshift nonsense mutation of MYH7 in the second case and a known mutation in the first case. The c.5059-5061delGAG variant occurs within a coiled coil region of the protein that is conserved across species. Furthermore, this in-frame deletion was not observed in approximately 6500 individuals of European and African American ancestry in the NHLBI Exome Sequencing Project, which indicates that it is not a common benign variant in these populations. Therefore, we interpret c.5059-5061delGAG as a pathogenic variant. According to the American College of Medical Genetics and Genomics, both mutations are pathogenetic [21]. The c.C5533T (p.R1845W) mutation alters the interactions between filaments so that their assembly is less constrained, causing the formation of abnormally large, degradation-resistant structures [22]. Tajsharghi et al. (2003) suggested that the mutation may interfere with the interaction of MYH7 with myosin-binding proteins and inhibit myosin assembly into thick filaments [23]. It is postulated that the phenotype depends on the location of the mutation in the MYH7 gene; mutations in the globular head, distal rod, and middle or proximal rod regions cause FHDCM, MSM, and LDM, respectively [5, 22]. Tail domain mutations are supposed to disrupt either myosin dimerization or interactions with other sarcomeric proteins, or both; at the same time, heart involvement is rare [24]. In contrast, myopathy with heart involvement for mutations in the globular head has been described [6].
Conclusion
The mutation of MYH7 may cause a spectrum of disease, which is, clinically and genetically, highly heterogeneous. The same phenotype can be attributed to several gene mutations, and the same gene mutations can lead to different clinical manifestations. We propose that the spectrum of diseases may be named as MYH7-related myopathy. Muscle biopsy has contributed to the understanding of pathological features: the exclusively specific one is the hyaline body. In our case, the patient presented as congenital core myopathy, indicating that the spectrum of pathogenetic genes of CNM should include MYH7.
References
Jaenicke T, Diederich KW, Haas W, Schleich J, Lichter P, Pfordt M, Bach A, Vosberg HP (1990) The complete sequence of the human beta-myosin heavy chain gene and a comparative analysis of its product. Genomics 8(2):194–206. https://doi.org/10.1016/0888-7543(90)90272-V
Walsh R, Rutland C, Thomas R, Loughna S (2010) Cardiomyopathy: a systematic review of disease-causing mutations in myosin heavy chain 7 and their phenotypic manifestations. Cardiology 115(1):49–60. https://doi.org/10.1159/000252808
Yüceyar N, Ayhan Ö, Karasoy H, Tolun A (2015) Homozygous MYH7 R1820W mutation results in recessive myosin storage myopathy: scapuloperoneal and respiratory weakness with dilated cardiomyopathy. Neuromuscul Disord 25(4):340–344. https://doi.org/10.1016/j.nmd.2015.01.007
Naddaf E and Waclawik, Andrew J. (2015) Two Families With MYH7 Distal Myopathy Associated With Cardiomyopathy and Core Formations. J Clin Neuromusc Dis 16(3):164–169 https://doi.org/10.1097/CND.0000000000000069
Muelas N, Hackman P, Luque H, Garces-Sanchez M, Azorin I, Suominen T, Sevilla T, Mayordomo F, Gomez L, Marti P, Maria Millan J, Udd B, Vilchez JJ (2010) MYH7 gene tail mutation causing myopathic profiles beyond Laing distal myopathy. Neurology 75(8):732–741. https://doi.org/10.1212/WNL.0b013e3181eee4d5
Overeem S, Schelhaas HJ, Blijham PJ, Grootscholten MI, ter Laak HJ, Timmermans J, van den Wijngaard A, Zwarts MJ (2007) Symptomatic distal myopathy with cardiomyopathy due to a MYH7 mutation. Neuromuscul Disord 17(6):490–493. https://doi.org/10.1016/j.nmd.2007.02.007
Laing NG, Ceuterick-de Groote C, Dye DE, Liyanage K, Duff RM, Dubois B, Robberecht W, Sciot R, Martin JJ, Goebel HH (2005) Myosin storage myopathy: slow skeletal myosin (MYH7) mutation in two isolated cases. Neurology 64(3):527–529. https://doi.org/10.1212/01.WNL.0000150581.37514.30
Shingde MV, Spring PJ, Maxwell A, Wills EJ, Harper CG, Dye DE, Laing NG, North KN (2006) Myosin storage (hyaline body) myopathy: a case report. Neuromuscul Disord 16(12):882–886. https://doi.org/10.1016/j.nmd.2006.09.001
Jie L et al (2015) Mutation and clinical relevance in a large cohort of unrelated Chinese patients with hypertrophic cardiomyopathy. Zhonghua Xin Xue Guan Bing Za Zhi 43(8):682–689
Liu W, Liu W, Hu D, Zhu T, Ma Z, Yang J, Xie W, Li C, Li L, Yang J, Li T, Bian H, Tong Q (2013) Mutation spectrum in a large cohort of unrelated Chinese patients with hypertrophic cardiomyopathy. Am J Cardiol 112(4):585–589. https://doi.org/10.1016/j.amjcard.2013.04.021
Park JM, Kim YJ, Yoo JH, Hong YB, Park JH, Koo H, Chung KW, Choi BO (2013) A novel MYH7 mutation with prominent paraspinal and proximal muscle involvement. Neuromuscul Disord 23(7):580–586. https://doi.org/10.1016/j.nmd.2013.04.003
Oda T, Xiong H, Kobayashi K, Wang S, Satake W, Jiao H, Yang Y, Cha PC, Hayashi YK, Nishino I, Suzuki Y, Sugano S, Wu X, Toda T (2015) A de novo mutation of the MYH7 gene in a large Chinese family with autosomal dominant myopathy. Human Genome Variation 2:15022. https://doi.org/10.1038/hgv.2015.22
Pegoraro E, Gavassini BF, Borsato C, Melacini P, Vianello A, Stramare R, Cenacchi G, Angelini C (2007) MYH7 gene mutation in myosin storage myopathy and scapulo-peroneal myopathy. Neuromuscul Disord 17(4):321–329. https://doi.org/10.1016/j.nmd.2007.01.010
Fiorillo C et al (2016) MYH7-related myopathies: clinical, histopathological and imaging findings in a cohort of Italian patients. Orphanet J Rare Dis 11(1):91. https://doi.org/10.1186/s13023-016-0476-1
Clarke NF, Amburgey K, Teener J, Camelo-Piragua S, Kesari A, Punetha J, Waddell LB, Davis M, Laing NG, Monnier N, North KN, Hoffman EP, Dowling JJ (2013) A novel mutation expands the genetic and clinical spectrum of MYH7-related myopathies. Neuromuscul Disord 23(5):432–436. https://doi.org/10.1016/j.nmd.2013.02.009
Cullup T, Lamont PJ, Cirak S, Damian MS, Wallefeld W, Gooding R, Tan SV, Sheehan J, Muntoni F, Abbs S, Sewry CA, Dubowitz V, Laing NG, Jungbluth H (2012) Mutations in MYH7 cause multi-minicore disease (MmD) with variable cardiac involvement. Neuromuscul Disord 22(12):1096–1104. https://doi.org/10.1016/j.nmd.2012.06.007
Olive M (2009) Extralysosomal protein degradation in myofibrillar myopathies. Brain Pathol 19(3):507–515. https://doi.org/10.1111/j.1750-3639.2009.00288.x
Ortolano S, Tarrío R, Blanco-Arias P, Teijeira S, Rodríguez-Trelles F, García-Murias M, Delague V, Lévy N, Fernández JM, Quintáns B, Millán BS, Carracedo Á, Navarro C, Sobrido MJ (2011) A novel MYH7 mutation links congenital fiber type disproportion and myosin storage myopathy. Neuromuscul Disord 21(4):254–262. https://doi.org/10.1016/j.nmd.2010.12.011
Lin P, Liu X, Zhao D, Dai T, Wu H, Gong Y, Yan C (2016) DNM2 mutations in Chinese Han patients with centronuclear myopathy. Neurol Sci 37(6):995–998. https://doi.org/10.1007/s10072-016-2513-1
Chen T, Pu C, Wang Q, Liu J, Mao Y, Shi Q (2015) Clinical, pathological, and genetic features of dynamin-2-related centronuclear myopathy in China. Neurol Sci 36(5):735–741. https://doi.org/10.1007/s10072-014-2028-6
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 17(5):405–423. https://doi.org/10.1038/gim.2015.30
Armel TZ, Leinwand LA (2009) Mutations in the beta-myosin rod cause myosin storage myopathy via multiple mechanisms. Proc Natl Acad Sci U S A 106(15):6291–6296. https://doi.org/10.1073/pnas.0900107106
Tajsharghi H, Thornell LE, Lindberg C, Lindvall B, Henriksson KG, Oldfors A (2003) Myosin storage myopathy associated with a heterozygous missense mutation in MYH7. Ann Neurol 54(4):494–500. https://doi.org/10.1002/ana.10693
Meredith C, Herrmann R, Parry C, Liyanage K, Dye DE, Durling HJ, Duff RM, Beckman K, de Visser M, van der Graaff MM, Hedera P, Fink JK, Petty EM, Lamont P, Fabian V, Bridges L, Voit T, Mastaglia FL, Laing NG (2004) Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause laing early-onset distal myopathy (MPD1). Am J Hum Genet 75(4):703–708. https://doi.org/10.1086/424760
Acknowledgements
We wish to thank the people with myopathy who participated in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study was approved by the Ethical Committee of the Third Hospital of Hebei Medical University and was conducted in accordance with the Declaration of Helsinki.
Conflict of interest
The authors declare that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Li, N., Zhao, Z., Shen, H. et al. MYH7 mutation associated with two phenotypes of myopathy. Neurol Sci 39, 333–339 (2018). https://doi.org/10.1007/s10072-017-3192-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-017-3192-2