
Abstract
Neuropsychological examinations in myotonic dystrophy (DM) patients show a great variability of results from a condition of intellectual disability to the subtle cognitive impairments. It is unclear if different clusters of neuropsychological deficits appear in different phenotypes of DM, or if there are patients with no cognitive deficit at all. The aim of this study is to assess cognitive impairments among patients with different phenotypes of DM type 1 (DM1) and type 2 (DM2), and to potentially define cognitive clusters in these disorders. Study comprised 101 DM1 and 46 DM2 adult patients who were genetically confirmed. Patients underwent analysis of five cognitive domains (visuospatial, executive, attention, memory and language). Virtually all DM1 patients had cognitive defect with approximately 2–3 cognitive domains affected. On the other hand, one-third of DM2 patients had completely normal neuropsychological findings, and in other two-thirds approximately 1–2 domains were affected. Cluster analysis showed that in both diseases visuospatial and executive dysfunctions seemed to be the main cognitive defects, while memory and language impairments appeared in more severe phenotypes. Our results showed that a single form of DM1 or DM2 may consist of several cognitive clusters. Understanding of cognitive impairments in DM is very important to follow positive and side effects in ongoing and future clinical trials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Myotonic dystrophies (DM) are autosomal dominant multisystemic disorders that affect various tissues, including the brain [1]. Pathological expansion of CTG repeats in DM1 and CCTG repeats in DM2 cause impaired alternative splicing of pre-messenger RNAs for various proteins, including the TAU and NMDA [2]. Brain tissue from patients with DM1 and DM2 lack TAU-immunoreactivity neurofibrillary tangles (NFTs) containing TAU-E2 and TAU-E3 [3]. Other molecular mechanisms may also be responsible for CNS symptoms in DM.
Neuroimaging studies on DM1 patients showed reduction of the brain gray and white matter volume, white matter hyperintense lesions and microstructural changes of the white matter [4–7]. Previous studies showed less pronounced gray matter loss in DM2 brain, while white matter was similarly impaired in both diseases [4–6]. It seems that white matter tracts’ damage and disruption of complex neuronal networks are responsible for cognitive and behavioral symptoms in DM [7, 8] and recently it has been showed that alteration of brain connectomics fit well with the cognitive deficits [9].
Cognitive impairments in DM1 depend on the age at onset of the disease. Congenital patients have mental retardation, while those with onset in the childhood have learning disabilities due to lower IQ, the attention deficit/hyperactivity disorder, autistic behavior, visuospatial impairment, lack of interest and inhibition [10]. Adult-onset DM1 patients have dysexecutive syndrome and visuospatial deficit, while verbal functions and memory seem to be age dependent [11–14]. Late-onset DM1 patients were reported to have predominantly age-dependent memory deficit [13].
Cognitive involvement is less pronounced in DM2. Main findings are executive dysfunction and certain degree of episodic memory impairment [11–15]. Congenital and childhood phenotypes of DM2 as well as mental retardation were not described. There were no comparisons of cognitive findings regarding age at onset of DM2.
Neuropsychological examinations in DM patients show a great variability of results from a condition of intellectual disability to the subtle impairments [6, 14]. Thus, reported results for a group as a whole are not applicable for individual cases. It is also unclear if different clusters of neuropsychological deficits appear, or if there are DM patients with no cognitive deficit at all. Axford et al. [16] reported that definition of affected cognitive domains is needed for different groups of DM patients.
The aim of this study was to assess cognitive impairments among patients with different phenotypes of DM1 and DM2, and to potentially define cognitive clusters in these disorders.
Patients and method
This retrospective study comprised 101 DM1 and 46 DM2 adult patients that were tested at the Department for Neuropsychology of the Neurology Clinic, Clinical Centre of Serbia at the University of Belgrade from January, 2007 until December, 2014. Approval was received from the Ethics Committee of the School of Medicine, University of Belgrade. All patients signed informed consent.
Clinical and electrophysiological diagnosis in all patients was confirmed by triplet repeat primed polymerase chain reaction (TR-PCR) [17]. In both DM1 and DM2 patients, age at onset was determined by the structured clinical interview with patients. DM1 patients were divided into four phenotypes: childhood-onset DM1 (cDM1) with age at onset from one to ten years, juvenile-onset DM1 (jDM1) with age at onset from ten to 20 years, adult-onset DM1 (aDM1) with age at onset between 20 and 40 years, and finally late DM1 (lDM1) with onset after age of 40. Patients with congenital DM1 were excluded from the study. We divided DM2 patients into two phenotypes: adult-onset DM2 (aDM2) with age at onset between 20 and 40 years and late DM2 (lDM2) with onset after age of 40. This division was not previously used, but we hypothesized it is appropriate due to the similarities between DM1 and DM2.
Severity of muscular involvement in DM1 was analyzed using the Muscular Impairment Rating Scale (MIRS) [18]. Since MIRS is not applicable in DM2, we added strength of the weakest muscle of proximal/distal muscle groups of the arms/legs [0–5 scale according to the Medical Research Council (MRC) scale], with maximum score being 20. Following muscle groups were tested: shoulder abductors and adductors, elbow flexors and extensors, wrist and finger flexor and extensors, hip flexors, extensors, abductors and adductors, knee flexors and extensors, plantar and dorsal ankle and toe flexors. Muscle strength was documented based on manual muscle testing by neurologist (V.R.S).
Patients underwent analysis of five cognitive domains (visuospatial, executive, attention, memory and language) performed by experienced neuropsychologists (V.I., A.P). Testing was performed in the morning after breakfast and lasted for approximately one hour and a half. Raven Standard Progressive Matrices (RSPM) [19] were used as a measure of general intellectual level. Global cognitive status was assessed using the Mini Mental State Examination (MMSE) and Addenbrooke’s Cognitive Examination-Revised (ACE-R) [20]. Digit Span and Block Design subtests from the Serbian version of the Wechsler Adult Intelligence Scale-Revised (WAIS-R) were used, and scaled scores below six were considered abnormal [21]. For other neuropsychological tests, results were considered abnormal if they were one standard deviation (SD) below or above the mean value of healthy population according to the following references: the Rey Auditory Verbal Learning Test (RAVLT) [22], copy and recall of the Rey–Osterrieth Complex Figure (ROCF) [23], Trail Making Test A and B (TMT-A and TMT-B) [23], Wisconsin Card Sorting Test (WCST) (number of achieved categories) [23], the Boston Naming Test (BNT) [24] and the semantic verbal fluency (SF) [21]. Visuospatial deficit was defined as an achievement of one SD below norm on both Block Design and copy of ROCF tests, or two SDs below norm on any of these. Executive dysfunction was defined as follows: scores one SD below norm on both WCST and TMT-B, or two SDs below norm on any of these. Attention deficit was defined as an achievement one SD below norm on both Digit Span and TMT-A, or two SDs below norm on any of these tests. Memory impairment was considered if patients scored one SD below norm on both copy of ROCF and RAVLT recognition, or two SDs below norm on any of these tests. Impaired language function was defined as a result one SD below norm on both BNT and CF tests, or two SDs below norm on any of these tests.
Normality of data was tested by the Kolmogorov–Smirnov test. χ 2 Test, Mann–Whitney U test, Kruskal–Wallis test, Student t test and ANOVA were used to compare sociodemographic and clinical parameters between patient groups, as appropriate. We did not compare raw results on each neuropsychological test since norms depend on gender, age and education, and patient groups were not matched for these parameters. Thus, χ 2 test was used to assess differences in the number of subjects that were one and two SDs below norm on each test and to compare proportions of patients with affection of each psychological domain. Hierarchical cluster analysis was applied to form groups with impairment of similar cognitive domains within each form of the diseases. Significant testing was two sided, and level of statistical significance of 0.05 was corrected for multiple comparisons.
Results
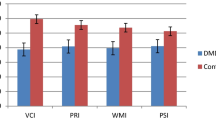
Main sociodemographic and clinical features of investigated patients are presented in Tables 1 and 2. Results of neuropsychological testing are given in Table 3 and Fig. 1. Patients with DM2 had significantly better results on RSPM, as a measure of general intellectual level. ACE-R results were the worst in patients with aDM1 and lDM1.

Results on neuropsychological testing in different phenotypes of DM1 and DM2. Results are shown as a percentage of patients with cognitive deficit in each domain (visuospatial, executive, attention, memory and language). *p < 0.05 for difference between patient groups in each cognitive domain
Number of impaired cognitive domains was less in DM2 (range 1–2) than in DM1 (range 2–4). Visuospatial dysfunction was more common in DM1 patients (>75%) but also present in approximately half of DM2 patients. Executive dysfunction was also more common in DM1. Attention deficit was the least common impairment in all DM1 groups. Memory impairment was the most prominent in patients with lDM1. The same applied to language dysfunction, although this was not of statistical significance.
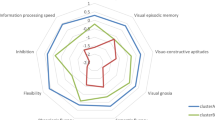
Results of cluster analysis are shown in Fig. 2. We identified two cognitive clusters among patients with cDM1: cluster A with predominant visuospatial impairment and more severe cluster B with visuospatial, executive, memory and language deficits. These two clusters did not differ in any of the sociodemographic and clinical parameters. In patients with jDM1 three clusters were found: A with executive/attentional deficit, B with visuospatial impairment, and C with both executive and visuospatial deficit as well as possible affection of memory and language. Differences in sociodemographic and clinical parameters were not observed between these three clusters. In aDM1 patients, we also identified three clusters: A with dominant visuospatial impairment, B mostly with language impairment, and cluster C visuospatial and memory impairment. Patients in cluster A were younger than patients in clusters B and C (37.8 ± 9.2 vs. 47.6 ± 8.1 vs. 48.7 ± 6.3 years, p = 0.003). aDM1 patients in cluster A also had shorter duration of disease than patients in clusters B and C (10.0 ± 6.1, 21.3 ± 8.4, 20.9 ± 7.9 years, p = 0.003). All of our patients with lDM1 were grouped into the same cluster with affection of almost all domains.

Clusters of cognitive profiles among different phenotypes of DM1 and DM2. V visuospatial function, E executive functions, A attention, M memory, L language. Gray rectangle impairment of corresponding cognitive domain. Patients with aDM2 were clustered as follows: A normal neuropsychological findings, B visuospatial impairment, C affection of multiple cognitive domains. lDM2 patients were grouped into two clusters: A normal, B multiple domains affection. Clusters did not differ regarding sociodemographic and clinical features neither in aDM2, nor in lDM2
Regarding DM2 adult phenotype, patients were clustered as follows: normal cognitive findings, visuospatial impairment, and multiple domains affection. Late DM2 were grouped in normal cluster and multiple domain impairment. Clusters did not differ regarding sociodemographic and clinical features neither in aDM2, nor in lDM2.
Discussion
In this study, we intended to organize heterogeneous pattern of cognitive impairment in DM1 and DM2, since this was previously suggested as necessary by experts in the field [16, 25].
Virtually all our DM1 patients had certain cognitive defect with approximately 2–3 cognitive domains affected. Visuospatial impairment seems to be the main cognitive defect with prevalence of 75–86% among different DM1 phenotypes. This is in accordance with previous results that showed poorer achievement in DM1 patients compared to HCs, with half or more patients having results below norms [12, 13, 25, 26]. Visuospatial dysfunction might have a significant influence on patient’s everyday life including spacial orientation, driving, reading maps, inability to perform jobs that requests construction, etc. [11]. In line with this, we previously found that visuospatial defect significantly influenced quality of life of jDM1 patients [27].
Executive dysfunction was the second most common cognitive impairment in DM1 with 60–79% of our patients being affected. Accordingly, previous studies reported worse results on WCST in DM1 patients compared to HCs, with approximately two-thirds of patients having scores below norms [11, 25], while results on TMT-B were below norm in 33–87% of DM1 subjects depending on the study [13]. It is of note that executive dysfunction in our cohort was usually a part of a more severe cognitive cluster in cDM1, jDM1 and aDM1, and often associated with certain level of visuospatial impairment. Thus, it is possible that bad achievement on executive function tests might be at least partially explained by visuospatial defect. Visuospatial defect suggests affection of the right hemisphere, especially parietal lobe which has very well developed links with other lobes including prefrontal, which anatomically might explain influence of visuospatial impairment on executive dysfunction. Executive dysfunction reduces initiative, planning abilities and decision making, leads to apathy and inactivity which may have significant influence on quality of life in DM1 patients. This was confirmed in a paper by Antonini et al. [28]. More recently, a reduced disease awareness which interferes with different physical and life domains has also been shown [29].
Memory and language impairments were observed almost exclusively in more severe clusters among cDM1, jDM1 and aDM1 patients. aDM1 subjects with older age and longer duration of disease were more susceptible to memory and language defects which suggest that these impairments are probably caused by degenerative process, vascular and/or metabolic dysfunction. Furthermore, patients with lDM1, as the oldest group, had the affection of multiple cognitive domains including the highest prevalence of memory impairment (more than 80%). In our study, these patients had normal general intellectual level which suggests normal brain development. However, they scored very poor on cognitive screening tests that are tools for identification of mild cognitive impairment and dementia, as well as on all tests for specific cognitive domains. This is in accordance with previous research that showed memory and naming function decline during time in DM1 subjects [30, 31]. Furthermore, Sansone et al. showed age-dependent memory deficit suggesting dementia in patients with lDM1 with only 50–150 CTG repeats [31]. A recent paper indicated that DM1 patients scored cognitively worse at 5-year follow-up compared to baseline which was associated with a younger age at onset and longer duration of disease [32]. Altogether, these findings support a possible degenerative brain processes in DM1.
In our study, there were no differences in cognitive achievements between aDM2 and lDM2, thus our division on these two phenotypes seems to be inappropriate. Patients with DM2 had generally better neuropsychological results in comparison to DM1 subjects. General intellectual level was lower than normal in only about 8% of DM2 patients. Screening tests for dementia showed cognitive decline in up to 25% of them. This is in accordance with previous studies that reported normal general intellectual level in DM2 and less than one-fourth of them with cognitive decline diagnosed with dementia screening tests [10, 14].
Number of affected cognitive domains in DM2 was less than in DM1. There are only few studies that investigated neuropsychological performance in DM2 so far, and all of them included small number of patients (between 9 and 22) [4–6, 11, 12, 15, 30]. Some authors stated that described neuropsychological impairment in DM2 is subtle and not clinically relevant [5, 12]. Our previous research demonstrated lesser degree of cognitive impairment in 34 DM2 patients compared to the same number of age- and gender-matched aDM1 subjects [14]. Cluster analysis showed that not all patients with DM2 have cognitive impairment, but even one-third of them had completely normal neuropsychological findings.
Similar to DM1, visuospatial impairment seems to be the main cognitive defect in DM2 patients, with prevalence of approximately 50%. Executive dysfunction was the second most common cognitive deficit in DM2 with 32–43% of our patients affected, and it was usually a part of a more severe cognitive cluster in DM2, and almost always associated with a certain level of visuospatial impairment. Both visuospatial and executive dysfunctions were less common in DM2 than in DM1 which is in accordance with previous studies [5, 6, 14, 30]. However, intradimensional/extradimensional set shifting test showed similar results in both DM1 and DM2 suggesting a significant dysfunction of frontostriatal circuits [14].
Memory and language deficits were less common in DM2 than in DM1 and usually present in a more severe cognitive cluster. Contrary to DM1, verbal memory was impaired more than visuospatial. Memory deficit has been previously described in DM2 patients [4, 14, 30] signaling that besides frontal lobes, temporal lobes and hippocampi are also affected.
Our results showed that a single phenotype of DM1 or DM2 may consist of several cognitive clusters. Furthermore, these clusters may be similar among different phenotypes. It means that some other factors besides age at onset of disease might have influence on cognitive achievement. We failed to identify sociodemographic and clinical parameters that can differentiate between clusters, except for age in a DM1. A large cross-sectional study from the DM-scope nationwide registry has recently shown that gender is a modifying factor influencing DM1 phenotype severity and mortality [33]. Other factors may also be responsible: genes close to DMPK gene (DMWD, SIX5), gene modifiers, repeat-associated non-ATG (RAN) translation, microRNA, micro- and macrostructural brain changes, metabolic factors, environmental factors, etc.
Influence of central manifestations on quality of life was demonstrated in DM1 [27, 28], but this issue remains uninvestigated in DM2. We suppose that cognitive deficit might cause poorer education, worse working position and salary, and consequently worse social participation.
Our research has several limitations. Although all patients were genetically confirmed using RP-PCR, Southern blot with repeats number was not performed in all of them. Furthermore, this analysis was not always performed at the same moment when neuropsychological testing, thus we did not include this factor in the analysis. The second limitation is that we divided patients into phenotypes based on age at onset only, while some authors defined lDM1 using age at onset, muscular weakness and CTG repeats number. Relatively pronounced muscular impairment in our lDM1 patients may be partly explained by a long duration of disease in this subgroup. Another limitation is represented by a difference in gender frequency in DM1 and DM2 groups (54 vs. 28% males, respectively). This could be an important bias, because already mentioned data from the large French national registry showed that the sex is a modifying factor, at least for DM1. In particular, men had more cognitive impairment, lower educational level and they more commonly work in a specialized environment. In addition, males were more affected in their social and economic life compared to women [33].
Significant limitation of the study is a lack of neuroimaging data. This is mostly due to the retrospective nature of our research. Majority of analyzed patients were neuropsychologically tested as a regular part of clinical investigation of DM1 patients in our hospital. However, subgroups of subjects were analyzed as a part of different research protocols including neuroimaging, and these data were previously published. Our recent study using magnetic resonance imaging showed correlation between cognitive deficits and white matter involvement in DM1 (including hyperintensities and microstructural damage) [7]. Another research group also showed significant associations between structural and functional brain changes. Correlations of morphological MRI results with clinical findings were found for reduced flexibility of thinking and atrophy of the left secondary visual cortex in DM1 and of distinct subcortical brain structures in DM2 [8]. In addition, in DM2, depression was associated with brainstem atrophy, while daytime sleepiness correlated with volume decrease in the middle cerebellar peduncles, pons/midbrain and the right medio-frontal cortex [8]. Furthermore, transcranial sonography showed correlation of brainstem raphe hypoechogenicity with depression and fatigue in our DM1 patients, and with fatigue and excessive daytime sleepiness in our DM2 patients [34, 35]. Accordingly, Krogias and colleagues found a relation between mesencephalic raphe echogenicity and excessive daytime sleepiness in their DM patients [36]. Studies on correlation between neuroimaging data and cognitive performances are still lacking in DM1, DM2, and especially comparing DM1 and DM2, thus further research in this field is welcome.
Conclusions
Virtually all DM1 patients have cognitive defect with approximately two to three cognitive domains affected. On the other hand, one-third of DM2 patients have completely normal neuropsychological findings, and in other two-thirds of patients approximately one or two domains are affected. In both diseases, visuospatial and executive dysfunctions seem to be the main cognitive defects, while memory and language defects appear in more severe phenotypes. Our results showed that a single form of DM1 or DM2 may consist of several cognitive clusters. Understanding of these impairments is very important to follow positive and side effects in ongoing and future clinical trials.
References
Udd B, Krahe R (2012) The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol 11(10):891–905
de León MB, Cisneros B (2008) Myotonic dystrophy 1 in the nervous system: from the clinic to molecular mechanisms. J Neurosci Res 86(1):18–26
Maurage CA, Udd B, Ruchoux MM, Vermersch P, Kalimo H, Krahe R, Delacourte A, Sergeant N (2005) Similar brain tau pathology in DM2/PROMM and DM1/Steinert disease. Neurology 65(10):1636–1638
Weber YG, Roebling R, Kassubek J, Hoffmann S, Rosenbohm A, Wolf M, Steinbach P, Jurkat-Rott K, Walter H, Reske SN, Lehmann-Horn F, Mottaghy FM, Lerche H (2010) Comparative analysis of brain structure, metabolism, and cognition in myotonic dystrophy 1 and 2. Neurology 74(14):1108–1117
Minnerop M, Weber B, Schoene-Bake JC, Roeske S, Mirbach S, Anspach C, Schneider-Gold C, Betz RC, Helmstaedter C, Tittgemeyer M, Klockgether T, Kornblum C (2011) The brain in myotonic dystrophy 1 and 2: evidence for a predominant white matter disease. Brain 134(12):3530–3546
Romeo V, Pegoraro E, Ferrati C, Squarzanti F, Sorarù G, Palmieri A, Zucchetta P, Antunovic L, Bonifazi E, Novelli G, Trevisan CP, Ermani M, Manara R, Angelini C (2010) Brain involvement in myotonic dystrophies: neuroimaging and neuropsychological comparative study in DM1 and DM2. J Neurol 257(8):1246–1255
Caso F, Agosta F, Peric S, Rakočević-Stojanović V, Copetti M, Kostic VS, Filippi M (2014) Cognitive impairment in myotonic dystrophy type 1 is associated with white matter damage. PLoS One 9(8):e104697
Schneider-Gold C, Bellenberg B, Prehn C, Krogias C, Schneider R, Klein J, Gold R, Lukas C (2015) Cortical and subcortical grey and white matter atrophy in myotonic dystrophies type 1 and 2 is associated with cognitive impairment, depression and daytime sleepiness. PLoS One 10(6):e0130352
Serra L, Mancini M, Silvestri G, Petrucci A, Masciullo M, Spanò B, Torso M, Mastropasqua C, Giacanelli M, Caltagirone C, Cercignani M, Meola G, Bozzali M (2016) Brain connectomics’ modification to clarify motor and nonmotor features of myotonic dystrophy type 1. Neural Plast 2016:2696085
Meola G, Sansone V (2007) Cerebral involvement in myotonic dystrophies. Muscle Nerve 36:294–306
Meola G, Sansone V, Perani D, Scarone S, Cappa S, Dragoni C, Cattaneo E, Cotelli M, Gobbo C, Fazio F, Siciliano G, Mancuso M, Vitelli E, Zhang S, Krahe R, Moxley RT (2003) Executive dysfunction and avoidant personality trait in myotonic dystrophy type 1 (DM-1) and in proximal myotonic myopathy (PROMM/DM-2). Neuromuscul Disord 13(10):813–821
Gaul C, Schmidt T, Windisch G, Wieser T, Müller T, Vielhaber S, Zierz S, Leplow B (2006) Subtle cognitive dysfunction in adult onset myotonic dystrophy type 1 (DM1) and type 2 (DM2). Neurology 67(2):350–352
Modoni A, Silvestri G, Pomponi MG, Mangiola F, Tonali PA, Marra C (2004) Characterization of the pattern of cognitive impairment in myotonic dystrophy type 1. Arch Neurol 61:1943–1947
Peric S, Mandic-Stojmenovic G, Stefanova E, Savic-Pavicevic D, Pesovic J, Ilic V, Dobricic V, Basta I, Lavrnic D, Rakocevic-Stojanovic V (2015) Frontostriatal dysexecutive syndrome: a core cognitive feature of myotonic dystrophy type 2. J Neurol 262(1):142–148
Meola G, Sansone V, Perani D, Colleluori A, Cappa S, Cotelli M, Fazio F, Thornton CA, Moxley RT (1999) Reduced cerebral blood flow and impaired visual-spatial function in proximal myotonic myopathy. Neurology 53(5):1042–1050
Axford MM, Pearson CE (2013) Illuminating CNS and cognitive issues in myotonic dystrophy: Workshop report. Neuromuscul Disord 23(4):370–374
Radvansky J, Ficek A, Kadasi L (2011) Repeat-primed polymerase chain reaction in myotonic dystrophy type 2 testing. Genet Test Mol Biomark 15(3):133–136
Mathieu J, Boivin H, Meunier D, Gaudreault M, Begin P (2001) Assessment of a disease-specific Muscular Impairment Rating Scale in myotonic dystrophy. Neurology 56:336–340
Burke H (1985) Raven progressive matrices: more on norms, reliability, and validity. J Clin Psychol 41:231–235
Mioshi E, Dawson K, Mitchell J, Arnold R, Hodges JR (2006) The Addenbrooke’s cognitive examination revised (ACE-R): a brief cognitive test battery for dementia screening. Int J Geriatr Psychiatry 21:1078–1085
Pavlovic D (2003) Dijagnosticki testovi u neuropsihologiji, II edn. Grafos, Beograd
Geffen G, Hoar KJ, O’Hanlon AP, Clark CR, Geffen LB (1990) Performance measures of 16- to 86-year-old males and females on the auditory verbal learning test. Clin Neuropsychol 4:45–63
Spreen O, Strauss E (1991) A compendium of neuropsychological tests. Administration, norms and commentary. Oxford University Press, New York
Goodglass H, Kaplan E (1983) The assessment of aphasia and related disorders. Lea and Febiger, Philadelphia
Winblad S, Lindberg C, Hansen S (2006) Cognitive deficits and CTG repeat expansion size in classical myotonic dystrophy type 1 (DM1). Behav Brain Funct 2:16
Serra L, Silvestri G, Petrucci A, Basile B, Masciullo M, Makovac E, Torso M, Spanò B, Mastropasqua C, Harrison NA, Bianchi ML, Giacanelli M, Caltagirone C, Cercignani M, Bozzali M (2014) Abnormal functional brain connectivity and personality traits in myotonic dystrophy type 1. JAMA Neurol 71(5):603–611
Rakocevic-Stojanovic V, Peric S, Madzarevic R, Dobricic V, Ralic V, Ilic V, Basta I, Nikolic A, Stefanova E (2014) Significant impact of behavioral and cognitive impairment on quality of life in patients with myotonic dystrophy type 1. Clin Neurol Neurosurg 126C:76–81
Antonini G, Soscia F, Giubilei F, De Carolis A, Gragnani F, Morino S, Ruberto A, Tatarelli R (2006) Health-related quality of life in myotonic dystrophy type 1 and its relationship with cognitive and emotional functioning. J Rehabil Med 38(3):181–185
Baldanzi S, Bevilacqua F, Lorio R, Volpi L, Simoncini C, Petrucci A, Cosottini M, Massimetti G, Tognoni G, Ricci G, Angelini C, Siciliano G (2016) Disease awareness in myotonic dystrophy type 1: an observational cross-sectional study. Orphanet J Rare Dis 11:34
Modoni A, Silvestri G, Vita MG, Quaranta D, Tonali PA, Marra C (2008) Cognitive impairment in myotonic dystrophy type 1 (DM1): a longitudinal follow-up study. J Neurol 255(11):1737–1742
Sansone V, Gandossini S, Cotelli M, Calabria M, Zanetti O, Meola G (2007) Cognitive impairment in adult myotonic dystrophies: a longitudinal study. Neurol Sci 28(1):9–15
Winblad S, Samuelsson L, Lindberg C, Meola G (2016) Cognition in myotonic dystrophy type 1: a 5-year follow-up study. Eur J Neurol. doi:10.1111/ene
Dogan C, De Antonio M, Hamroun D, Varet H, Fabbro M, Rougier F, Amarof K, Arne Bes MC, Bedat-Millet AL, Behin A, Bellance R, Bouhour F, Boutte C, Boyer F, Campana-Salort E, Chapon F, Cintas P, Desnuelle C, Deschamps R, Drouin-Garraud V, Ferrer X, Gervais-Bernard H, Ghorab K, Laforet P, Magot A, Magy L, Menard D, Minot MC, Nadaj-Pakleza A, Pellieux S, Pereon Y, Preudhomme M, Pouget J, Sacconi S, Sole G, Stojkovich T, Tiffreau V, Urtizberea A, Vial C, Zagnoli F, Caranhac G, Bourlier C, Riviere G, Geille A, Gherardi RK, Eymard B, Puymirat J, Katsahian S, Bassez G (2016) Gender as a modifying factor influencing myotonic dystrophy type 1 phenotype severity and mortality: a Nationwide multiple databases cross-sectional observational study. PLoS One 11(2):e0148264
Peric S, Pavlovic A, Ralic V, Dobricic V, Basta I, Lavrnic D, Rakocevic Stojanovic V (2014) Transcranial sonography in patients with myotonic dystrophy type 1. Muscle Nerve 50(2):278–282
Rakocevic-Stojanovic V, Peric S, Savic-Pavicevic D, Pesovic J, Mesaros S, Lavrnic D, Jovanovic Z, Pavlovic A (2016) Brain sonography insight into the midbrain in myotonic dystrophy type 2. Muscle Nerve 53(5):700–704
Krogias C, Bellenberg B, Prehn C, Schneider R, Meves SH, Gold R, Lukas C, Schneider-Gold C (2015) Evaluation of CNS involvement in myotonic dystrophy type 1 and type 2 by transcranial sonography. J Neurol 262(2):365–374
Acknowledgements
This study was supported by the Ministry of Education, Science and Technological Development of Serbia (Grant #175083 to VRS) and by FMM—Fondazione Malattie Miotoniche, Italy (to GM).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Peric, Dr. Dobricic, Mr. Pesovic, Dr. Novakovic, Dr. Savic-Pavicevic, Dr. Mijajlovic and Dr. Rakocevic Stojanovic report grants from the Ministry of Education, Science and Technological Development of Serbia. Dr. Meola received grant from the FMM-Fondazione Malattie Miotoniche, Italy. The authors declare that they have no other conflicts of interest.
Rights and permissions
About this article

Cite this article
Peric, S., Rakocevic Stojanovic, V., Mandic Stojmenovic, G. et al. Clusters of cognitive impairment among different phenotypes of myotonic dystrophy type 1 and type 2. Neurol Sci 38, 415–423 (2017). https://doi.org/10.1007/s10072-016-2778-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-016-2778-4