
Abstract
Primary familial brain calcification is a neuropsychiatric disorder with calcium deposits in the brain, especially in basal ganglia, cerebellum and subcortical white matter. The disease is characterized by a clinical heterogeneity, with a various combination of symptoms that include movement disorders and psychiatric disturbances; asymptomatic patients have been also reported. To date, three causative genes have been found: SLC20A2, PDGFRB and PDGFB. SLC20A2 gene codes for the ‘sodium-dependent phosphate transporter 2’ (PiT-2), a cell membrane transporters of inorganic phosphate, involved in Pi uptake by cells and maintenance of Pi body levels. Over 40 pathogenic variants of SLC20A2 have been reported, affecting the regulation of Pi homeostasis. It was hypothesized that SLC20A2 mutations cause brain calcification most likely through haploinsufficiency. PDGFRB encodes for the platelet-derived growth factor receptor-β (PDGFRβ), a cell-surface tyrosine-kinase (RTK) receptor that regulates cell proliferation, migration, survival and differentiation. PDGFB encodes for the ‘platelet-derived growth factor beta’ (PDGFβ), the ligand of PDGFRβ. The loss of function of PDGFRβ and PDGFβ could lead to the impairment of the pericytes function and blood brain barrier integrity, causing vascular and perivascular calcium accumulation. SLC20A2 accounts for about 40 % of familial form and 14 % of sporadic cases, while PDGFRB and PDGFB mutations are likely rare. However, approximately 50 % of patients are not genetically defined and there should be at least another causative gene.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
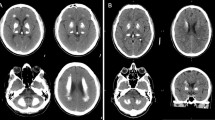
Primary familial brain calcification (PFBC), also called idiopathic basal ganglia calcification (IBGC), or Fahr’s disease, is a neuropsychiatric disorder characterized by bilateral brain calcification, usually transmitted as an autosomal dominant trait with incomplete penetrance [1–5]. The disease was first described by Delacour in 1850 [1] and recently, different genetic causes of PFBC have been discovered [2–4]. PFBC patients show bilateral calcium depositions commonly in basal ganglia, but other brain regions may be involved, such as the cerebellum, thalamus and subcortical white matter (Fig. 1) [5]. Furthermore, a mild frontal lobe atrophy with enlarged lateral ventricles and cerebellar atrophy might be present [6]. Neuropathological examinations showed accumulation of granular material, mostly calcium salts, around the walls of capillaries, small arteries and veins of the affected brain regions [5, 6]. PFBC patients display normal serum levels of calcium, phosphate, alkaline phosphatase and parathyroid hormone features which distinguish ‘primary’ brain calcification from those ‘secondary’ to parathyroid dysfunctions. Indeed, a common cause of basal ganglia calcification is hypoparathyroidism (HP): low serum levels of parathyroid hormone (PTH) could give rise to hypocalcaemia and hyperphosphatemia, causing an ectopic calcification in brain tissue [7]. Secondary brain calcification might occur also in several conditions, such as mitochondrial disorders (mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes—MELAS; myoclonic epilepsy associated with ragged red fibres—MERRF), autoimmune diseases (systemic lupus erythematosus), infectious and inflammation disorders. Furthermore, calcium deposition in the basal ganglia may be encountered in some neurodegenerative disorders (pantothenate kinase-associated neurodegeneration, PKAN; polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy, PLOSL) and congenital syndromes (Aicardi–Goutieres syndrome) [5, 8]. Last, brain calcifications of undefined pathogenicity are an incidental finding in about 1–20 % of brain CT scans of healthy people, especially elderly, as a consequence of the ageing process [9]. PFBC is clinically characterized by a wide spectrum of symptoms: movement disorders, cognitive impairment and psychiatric signs [5–10]. Movement disorders include akinetic-hypertonic syndrome with or without tremor, chorea, dystonia and oro-facial dyskinesia. Among cognitive impairment, the most common sign is memory loss; furthermore, a large number of patients present a dysexecutive syndrome. The main psychiatric signs are mood disorders, including depression and bipolar disorder, followed by psychosis. Moreover, seizures and chronic headache have been also reported [5, 10]. Patients can show a variable combination of these symptoms. The age of onset of the disease is typically between 30 and 60 years, but both the severity of symptoms and the age of onset are very variable. Furthermore, a large number of patients with brain calcification, about 30 %, can remain asymptomatic throughout life [5, 10]; therefore, sometimes the diagnosis of PFBC is based only on CT scan findings [5]. In addition, no correlation between the location, extent of calcification and the severity of symptoms has been observed [10]. The PFBC is usually inherited in an autosomal dominant fashion with incomplete clinical penetrance. Recent studies have shown that PFBC is genetically heterogeneous; so far, mutations in three genes (SLC20A2, PDGFRB, PDGFB) have been discovered as a cause of autosomal dominant forms of PFBC. Yet, mutations in these three genes account for only approximately 50 % of the cases [2–4, 11], suggesting that additional disease-causing genes remain to be identified. Very recently, a first autosomal recessive form of brain calcification has been reported [12]. The spectrum of PFBC-causing mutations is very wide, with a uniform and widespread distribution worldwide. So far, genotype–phenotype correlations have not been observed in patients with SLC20A2 and PDGFRB mutations [10].

Brain calcifications detected at CT scans of different patients. Arrows indicate calcifications in basal ganglia (a, b), brainstem and cerebellum (c)
Molecular genetics
SLC20A2
The SLC20A2 gene located on chromosome 8 (8p11.21) codes for the PiT-2 protein (652 amino acids), the ‘sodium-dependent phosphate transporter 2’. This protein belongs to the ‘SLC20-type III Na+ co-transporter family’, that also includes PiT-1 (sodium-dependent phosphate transporter 1), encoded by SLC20A1 [11]. PiT-1 and PiT-2 are cell membrane transporters of inorganic phosphate (Pi), consisting of 12 transmembrane domains, a large intracellular domain and extracellular N- and C- terminal tails [13, 14]. These transporters are widely expressed at various levels in human tissues and play a housekeeping role in Pi uptake by the cell and phosphate tissue homeostasis [13]. In 2012, Wang and colleagues [2] associated PFBC with PiT-2 by the discovery of seven mutations in the SLC20A2 gene (p.Gly498Arg, p.Ser601Gln, p.Ser601Leu, p.Glu575Lys, p.Thr595Met, p.Val42del, p.Pro470Leufs*37) in patients from seven PFBC families of various ancestry. All the missense variants and the deletion were used to perform 32Pi transport assays in Xenopus laevis oocytes. By co-expressing mutated and wild-type PiT-2 transporters, these Authors suggested that mutations in SLC20A2 most likely have an effect through haploinsufficiency [2]. In the mammalian brain, PiT-2 is ubiquitously expressed; at cellular level, PiT-2 has been mostly detected in neurons, astrocytes and endothelial cells and also in vascular smooth muscle cells (VSMCs) [15, 16]. Experiments with knockout mice for SLC20A2 gene confirmed the previously suggested hypothesis that calcification starts around the walls of brain vessels [2, 16]. SLC20A2 mutations might impair the Pi uptake, leading to a local increase in extracellular phosphate [16]. Afterwards, high levels of extracellular phosphate might result in a passive precipitation of calcium-phosphate products, triggering an active cell-mineralization process, probably via PiT-1 [2, 16]. Indeed, PiT-1 transporter is required for the normal bone cell differentiation and mineralization and is also involved in pathological smooth muscle cell (SMC) calcification. Interestingly, inducers of calcification, as the bone morphogenetic protein-2 (BMP-2), calcium and platelet-derived growth factor PDGF, upregulated PiT-1, but not PiT-2 expression [17, 18]. In vitro studies demonstrated that elevated phosphate levels result in the loss of smooth muscle markers (SM alpha actin, SM22 alpha) and expression of osteochondrogenic markers (Runx2/Cbfa1, osterix, alkaline phosphatase, osteopontin). Indeed, elevated phosphate concentrations may enhance the expression of the transcription factor Cbfa-1 (core-binding factor-1), that regulates the expression of osteogenic genes, such as osteopontin and osteocalcin [19]. Moreover, in vitro experiments showed that the phosphonoformic acid (PFA), an inhibitor of Pi transporters, abolishes the Pi uptake and prevents the mineralization of human aortic smooth muscle cells [20]. Although the involvement of PiT-1 in bone differentiation and cell mineralization has been deeply investigated, the molecular mechanism of calcification caused by PiT-2 transport dysfunctions is still to be elucidated and further studies are needed. So far, over 40 pathogenic variants in SLC20A2 gene have been reported in patients with PFBC, including missense, frameshift and non-sense mutations, but also deletions and one splice-site mutation; among them, one de novo variant has been found (Table 1) [21]. Missense mutations in PiT-2 protein could impair transport function; for instance, His502 and Glu575, substituted by a glutamine and a lysine, respectively, are critical for Pi transport [2, 14, 22]. Some frameshift and non-sense mutations, such as p.Leu170*, p.Val195Leufs*61, p.Pro470Leufs*37 and p.R172fs*19, are predicted to generate a premature termination codon (PTC) leading to the “non-sense mediated decay” (NMD) process, a surveillance mechanism that degrades aberrant mRNA, causing the loss of the transcript [22, 23]. Particularly, the frameshift mutation p.R172fs*19 leads to a 30 % reduction of the SLC20A2 mRNA expression, revealing that NMD process takes place and confirming haploinsufficiency as the most likely disease mechanism [23]. Furthermore, a genomic deletion of 563 kb in the chromosome 8 (g.42275321_42329908del), including SLC20A2, has been described in one large family. This deletion comprised seven genes (VDAC3, SLC20A2, C8ORF40, CHRNB3, CHRNA6, THAP1, RNF170) and partial deletion of HOOK3; interestingly, THAP1 is the causative gene of a familial form of dystonia (DYT16), indeed the patients displayed dystonia as main symptom [24]. In two studies, mutations in the SLC20A2 gene have been found in 41 % of the familial PFBC cases and 14 % of sporadic patients, showing that PiT-2 impairment is a frequent and widespread cause of primary brain calcification [22, 25].
PDGFRB
The PDGFRB gene is located on chromosome 5 (5q33.1) and encodes the platelet-derived growth factor receptor-β (PDGFRβ, 1106 amino acids), recognized by growth factor homodimers PDGF-BB and PDGF-DD [26]. PDGFRβ is a cell-surface tyrosine-kinase receptor, consisting of five extracellular immunoglobulin (Ig) loops and an intracellular tyrosine-kinase domain [26]. In the brain, it is expressed in neurons, VSMCs and pericytes [3, 26]. The binding of the ligand triggers the dimerization, autophosphorylation and activation of the PDGFRβ receptor, which, in turn, initiates the downstream signalling leading to cell proliferation, migration, survival and differentiation [3, 26]. Recently, four missense mutations in the PDGFRB gene have been reported in one PFBC family and three sporadic cases (Table 2). All mutations, p.Leu658Pro, p.Arg695Cys, p.Arg987Trp and p.Glu1071Val, cause the substitution of conserved amino acids and were predicted to be pathogenic. Cell culture experiments showed that variants in the tyrosine-kinase domain (from 562 to 953 aa) reduce the receptor levels and the autophosphorylation [27]. It has been demonstrated that the missense mutation p.Leu658Pro reduces the kinase activity, while p.Arg987Trp mutation causes a rapid degradation of the receptor and impairs the activation of STAT3, a transcription activator, blocking the downstream signalling. On the other hand, the mutation p.Glu1071Val does not affect the phosphorylation and the signalling and probably it might be a polymorphism [28]. It has been hypothesized that the loss of function of PDGFRβ could lead to the impairment of the blood brain barrier (BBB) integrity, causing vascular and perivascular calcium accumulation [3]. Moreover, a deficient PDGF-β signalling is highly damaging to VSMCs and pericytes, resulting in complete lack of pericytes or pericyte hypoplasia, endothelial hyperplasia, increased vessel diameter, increased vascular permeability and vessel instability [4, 27]. Alternatively, it has been suggested that mutations in PDGFRB gene might be activating mutations, impairing the PDGFRβ-PiT-1 signalling and inducing VSMCs mineralization. In VSMCs, the PDGFRβ pathway enhances the expression of PiT-1, increasing the abundance of the receptor in the endoplasmic reticulum membranes and stimulating the Pi uptake [29]. To date, few functional analyses have been carried out to clarify the molecular mechanism of PFBC due to PDGFRB mutations. Recently, PDGFRB mutations have been also found in patients with autosomal dominant infantile myofibromatosis, a disorder of mesenchymal proliferation, characterized by benign tumour of soft tissue in infancy and childhood [30, 31]. Two germinal mutations, c.1681C>T (p.Arg561Cys) and c.1978C>A (p.Pro660Thr), and one somatic mutation c.1998C>A (p.Asn666Lys) have been reported. The variant p.Arg561Cys is located outside the kinase domain and probably compromises the auto-inhibition of the receptor. While the amino acids Asn666 and Pro660 are located in the kinase domain, and probably the variant p.Asn666Lys may abolish the interaction with inhibitors, deregulating the kinase activity [30, 31].
PDGFB
PDGFB gene is located on chromosome 22 (22q13.1) and encodes for the ‘platelet-derived growth factor beta’ (PDGFβ-241 amino acids), the ligand of the PDGFRβ receptor. PDGFβ is an antiparallel disulphide-linked dimer, a paracrine factor synthesized and secreted by angiogenic endothelial cells, which acts on pericytes and VSMCs which in turn have PDGFRβ in the cell-surface membrane [26]. In these cells, the PDGF signalling promotes the proliferation and migration along the newly developing blood vessels [32]. So far, eight PDGFB mutations, including one de novo variant, have been found in eight PFBC patients [33] (Table 3). Three are missense mutations: p.Leu9Arg inserts a charged amino acid in the signal peptide that is essential for the protein export; p.Leu119Pro occurs in the receptor-binding loop and p.*242>Tyrext*89 substitutes the stop codon with a tyrosine, leading to an extension of 89 codons in the transcript [4]. Three non-sense mutations, p.Gln145*, p.Gln147* and p.Arg149*, are predicted to remove part of the protein [4, 33]. The mutation P.Met1? may replace the start methionine, but the consequences of this variant have not yet been clarified [4]. Finally, a large intragenic deletion (7.2-kb) within PDGFB has been found in a patient with brain calcification and leukoencephalopathy [34]. This deletion comprises exons 3, 4 and 5, which encode for receptor-binding sites and dimerization domains, and might result in a truncated, not functional protein [34]. Mutations in PDGFB are predicted to be ‘loss of function’ and the discovery of a partial PDGFB gene deletion confirms this hypothesis [34]. Furthermore, Keller and colleagues showed that mice deficient in PDGFβ develop age-related calcified nodules in the thalamus and midbrain, which are similar to the lesions observed in the PFBC patients. Moreover, a correlation between endothelial PDGFβ, but not neuronal PDGFβ, and brain calcification in mice has been described [4]. Mice expressing PDGFβ, that lacks the retention motif which is essential for the diffusion of the protein in the tissues interstitium, showed an alteration of the local concentration and bioavailability of PDGFβ and a reduction of pericyte recruitment. These data strongly support a correlation between brain calcification and BBB impairment, caused by pericyte deficiencies [4].
Brain calcification and ISG15 gene
Recently, homozygous mutations in ISG15 gene have been found in six young patients with brain calcification, from three families from China, Iran and Turkey [12, 35]. ISG15 gene (1p36.33) encodes for an interferon (IFN)-α/β—inducible-ubiquitin-like modifier involved in the innate immune response to viral infection. It acts by conjugation to a target protein (ISGylation) or as a free and unconjugated protein and it is a negative regulator of IFN α/β immunity [12, 35]. Three mutations have been discovered in this gene: c.379G>T (p.Glu127*), c.336_337insG (p.Leu114fs) and c. 163 C>T (p.Gln55*). All mutations are in homozygous state and lead to the lack of ISG15 protein and a subsequent increase of IFN-α/β immunity. Also the Aicardi–Goutieres syndrome and spondyloenchondromatosis (SPENCD), in which brain calcification is a common feature, have been associated with up-regulation of IFN-α/β immunity [12, 35, 36]. ISG15 mutations are also linked to Mendelian susceptibility to mycobacterial disease (MSMD), in which severe clinical disease occurs following infection with weakly virulent mycobacteria, due to an insufficient production of ISG15-dependent IFN-γ [35]. However, brain calcification disease caused by mutation in ISG15 gene is quite different from PFBC, since the involvement of the IFN-α/β immunity and the autosomal recessive inheritance.
Conclusion
The recent genetic discoveries point to abnormalities of PiT-2 transport and the PDGFβ/PDGFRβ pathway, leading to the accumulation of calcium salts in the brain. PiT-2, PDGFβ and PDGFRβ are widely expressed in human tissues, but calcifications occur only in the brain. Future investigations are warranted to understand the detailed molecular mechanism leading to PFBC. PFBC is a clinically heterogeneous disease and the wide spectrum of symptoms could make the diagnosis challenging. The recent findings of disease-causing mutations in three genes confirm the previously suggested genetic heterogeneity of PFBC [11], and it allows a molecular diagnosis to be made in several patients. However, the genetic defect remains currently unknown in about 50 % of the autosomal dominant PFBC cases, suggesting the existence of at least another genetic form. Future work will be focused on the identification of mutations in additional genes for PFBC.
References
Delancourt A (1850) Ossification des capillaires du cerveau. Annales de Medecine et Psychologie 2:458–461
Wang C, Li Y, Shi L, Ren J, Patti M, Wang T, de Oliveira JR, Sobrido MJ, Quintáns B, Baquero M, Cui X, Zhang XY, Wang L, Xu H, Wang J, Yao J, Dai X, Liu J, Zhang L, Ma H, Gao Y, Ma X, Feng S, Liu M, Wang QK, Forster IC, Zhang X, Liu JY (2012) Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nat Genet 44(3):254–256
Nicolas G, Pottier C, Maltete D, Coutant S, Rovelet-Lecrux A, Legallic S, Rousseau S, Vaschalde Y, Guyant-Maréchal L, Augustin J, Martinaud O, Defebvre L, Krystkowiak P, Pariente J, Clanet M, Labauge P, Ayrignac X, Lefaucheur R, Le Ber I, Frébourg T, Hannequin D, Campion D (2013) Mutation of the PDGFRB gene as a cause of idiopathic basal ganglia calcification. Neurology 80(2):181–187
Keller A, Westenberger A, Sobrido MJ, García-Murias M, Domingo A, Sears RL, Lemos RR, Ordoñez-Ugalde A, Nicolas G, da Cunha JE, Rushing EJ, Hugelshofer M, Wurnig MC, Kaech A, Reimann R, Lohmann K, Dobričić V, Carracedo A, Petrović I, Miyasaki JM, Abakumova I, Mäe MA, Raschperger E, Zatz M, Zschiedrich K, Klepper J, Spiteri E, Prieto JM, Navas I, Preuss M, Dering C, Janković M, Paucar M, Svenningsson P, Saliminejad K, Khorshid HR, Novaković I, Aguzzi A, Boss A, Le Ber I, Defer G, Hannequin D, Kostić VS, Campion D, Geschwind DH, Coppola G, Betsholtz C, Klein C, Oliveira JR (2013) Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice. Nat Genet 45(9):1077–1082
Manyam BV (2005) What is and what is not ‘Fahr’s disease’. Parkinsonism Relat Disord 11(2):73–80
Wider C, Dickson DW, Schweitzer KJ, Broderick DF, Wszolek ZK (2009) Familial idiopathic basal ganglia calcification: a challenging clinical-pathological correlation. J Neurol 256(5):839–842
Caudarella R, Vescini F, Buffa A, Francucci CM (2007) Hyperphosphatemia: effects on bone metabolism and cardiovascular risk. J Endocrinol Invest 30(6 Suppl):29–34
Baba Y, Broderick DF, Uitti RJ, Hutton ML, Wszolek ZK (2005) Heredofamilial brain calcinosis syndrome. Mayo Clin Proc 80(5):641–651
Yamada M, Asano T, Okamoto K, Hayashi Y, Kanematsu M, Hoshi H, Akaiwa Y, Shimohata T, Nishizawa M, Inuzuka T, Hozumi I (2013) High frequency of calcification in basal ganglia on brain computed tomography images in Japanese older adults. Geriatr Gerontol Int 13(3):706–710
Nicolas G, Pottier C, Charbonnier C, Guyant-Maréchal L, Le Ber I, Pariente J, Labauge P, Ayrignac X, Defebvre L, Maltête D, Martinaud O, Lefaucheur R, Guillin O, Wallon D, Chaumette B, Rondepierre P, Derache N, Fromager G, Schaeffer S, Krystkowiak P, Verny C, Jurici S, Sauvée M, Vérin M, Lebouvier T, Rouaud O, Thauvin-Robinet C, Rousseau S, Rovelet-Lecrux A, Frebourg T, Campion D, Hannequin D, French IBGC Study Group (2013) Phenotypic spectrum of probable and genetically-confirmed idiopathic basal ganglia calcification. Brain 136(Pt 11):3395–3407
Oliveira JR, Spiteri E, Sobrido MJ, Hopfer S, Klepper J, Voit T, Gilbert J, Wszolek ZK, Calne DB, Stoessl AJ, Hutton M, Manyam BV, Boller F, Baquero M, Geschwind DH (2004) Genetic heterogeneity in familial idiopathic basal ganglia calcification (Fahr disease). Neurology 63(11):2165–2167
Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, Volpi S, Li Z, Sanal O, Mansouri D, Tezcan I, Rice GI, Chen C, Mansouri N, Mahdaviani SA, Itan Y, Boisson B, Okada S, Zeng L, Wang X, Jiang H, Liu W, Han T, Liu D, Ma T, Wang B, Liu M, Liu JY, Wang QK, Yalnizoglu D, Radoshevich L, Uzé G, Gros P, Rozenberg F, Zhang SY, Jouanguy E, Bustamante J, García-Sastre A, Abel L, Lebon P, Notarangelo LD, Crow YJ, Boisson-Dupuis S, Casanova JL, Pellegrini S (2014) Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature 517(7532):89–93
Forster IC, Hernando N, Biber J, Murer H (2013) Phosphate transporters of the SLC20 and SLC34 families. Mol Aspects Med 34(2–3):386–395
Bøttger P, Pedersen L (2011) Mapping of the minimal inorganic phosphate transporting unit of human PiT2 suggests a structure universal to PiT-related proteins from all kingdoms of life. BMC Biochem 12:21
Inden M, Iriyama M, Takagi M, Kaneko M, Hozumi I (2013) Localization of type-III sodium-dependent phosphate transporter 2 in the mouse brain. Brain Res 1531:75–83
Jensen N, Schrøder HD, Hejbøl EK, Füchtbauer EM, Oliveira JRM, Pedersen L (2013) Loss of function of Slc20a2 associated with familial idiopathic basal ganglia calcification in humans causes brain calcifications in mice. J Mol Neurosci 51(3):994–999
Lau WL, Festing MH, Gachelli CM (2010) Phosphate and vascular calcification: emerging role of the sodium-dependent phosphate co-transporter Pit-1. Thromb Haemost 104(3):464–470
Crouthamel MH, Lau L, Leaf EM, Chavkin NW, Wallingford M, Peterson DF, Li X, Liu Y, Chin MT, Levi M, Giachelli CM (2013) Sodium-dependent phosphate cotransporters and phosphate-induced calcification of vascular smooth muscle cells redundant roles for PiT-1 and PiT-2. Arterioscler Thromb Vasc Biol 33:2625–2632
Li X, Giachelli CM (2007) Sodium-dependent phosphate cotransporters and vascular calcification. Curr Opin Nephrol Hypertens 16:325–328
Giachelli CM, Jono S, Shioi A, Nishizawa Y, Mori K, Morii H (2001) Vascular calcification and inorganic phosphate. Am J Kidney Dis 38(4 Suppl 1):S34–S37
Ferreira JB, Pimentel L, Keasey MP, Lemos RR, Santos LM, Oliveira MF, Santos S, Jensen N, Teixeira K, Pedersen L, Rocha CR, Dias da Silva MR, Oliveira JR (2014) First report of a de novo mutation at SLC20A2 in a patient with brain calcification. J Mol Neurosci 54(4):748–751
Hsu SC, Sears RL, Lemos RR, Quintans B, Huang A, Spiteri E, Nevarez L, Mamah C, Zatz M, Pierce KD, Fullerton JM, Adair JC, Berner JE, Bower M, Brodaty H, Carmona O, Dobricić V, Fogel BL, García-Estevez D, Goldman J, Goudreau JL, Hopfer S, Janković M, Jaumà S, Jen JC, Kirdlarp S, Klepper J, Kostić V, Lang AE, Linglart A, Maisenbacher MK, Manyam BV, Mazzoni P, Miedzybrodzka Z, Mitarnun W, Mitchell PB, Mueller J, Novaković I, Paucar M, Paulson H, Simpson SA, Svenningsson P, Tuite P, Vitek J, Wetchaphanphesat S, Williams C, Yang M, Schofield PR, de Oliveira JR, Sobrido MJ, Geschwind DH, Coppola G (2013) Mutations in SLC20A2 are a major cause of familial idiopathic basal ganglia calcification. Neurogenetics 14(1):11–22
Zhang Y, Guo X, Wu A (2013) Association between a novel mutation in SLC20A2 and familial idiopathic basal ganglia calcification. PLoS One 8(2):e57060
Baker M, Strongosky AJ, Sanchez-Contreras MY, Yang S, Ferguson W, Calne DB, Calne S, Stoessl AJ, Allanson JE, Broderick DF, Hutton ML, Dickson DW, Ross OA, Wszolek ZK, Rademakers R (2013) SLC20A2 and THAP1 deletion in familial basal ganglia calcification with dystonia. Neurogenetics 15(1):23–30
Chen WJ, Yao XP, Zhang QJ, Ni W, He J, Li HF, Zhao GX, Murong SX, Wang N, Wu ZY (2013) Novel SLC20A2 mutations identified in southern Chinese patients with idiopathic basal ganglia calcification. Gene 529(1):159–162
Andrae J, Gallini R, Betsholtz C (2008) Role of platelet-derived growth factors in physiology and medicine. Genes Dev 22(10):1276–1312
Sanchez-Contreras M, Baker MC, Finch NA, Nicholson A, Wojtas A, Wszolek ZK, Ross OA, Dickson DW, Rademakers R (2014) Genetic screening and functional characterization of PDGFRB mutations associated with basal ganglia calcification of unknown etiology. Hum Mutat 35(8):964–971
Arts FA, Velghe AI, Stevens M, Renauld JC, Essaghir A, Demoulin JB (2014) Idiopathic basal ganglia calcification-associated PDGFRB mutation impair the receptor signalling. J Cell Mol Med 19(1):239–248
Villa-Bellosta R, Levi M, Sorribas V (2009) Vascular smooth muscle cell calcification and SLC20 inorganic phosphate transporters: effects of PDGF, TNF-alpha, and Pi. Pflugers Arch 458(6):1151–1161
Cheung YH, Gayden T, Campeau PM, LeDuc CA, Russo D, Nguyen VH, Guo J, Qi M, Guan Y, Albrecht S, Moroz B, Eldin KW, Lu JT, Schwartzentruber J, Malkin D, Berghuis AM, Emil S, Gibbs RA, Burk DL, Vanstone M, Lee BH, Orchard D, Boycott KM, Chung WK, Jabado NA (2013) Recurrent PDGFRB mutation causes familial infantile myofibromatosis. Am J Hum Genet 92(6):996–1000
Martignetti JA, Tian L, Li D, Ramirez MC, Camacho-Vanegas O, Camacho SC, Guo Y, Zand DJ, Bernstein AM, Masur SK, Kim CE, Otieno FG, Hou C, Abdel-Magid N, Tweddale B, Metry D, Fournet JC, Papp E, McPherson EW, Zabel C, Vaksmann G, Morisot C, Keating B, Sleiman PM, Cleveland JA, Everman DB, Zackai E, Hakonarson H (2013) Mutations in PDGFRB cause autosomal dominant infantile myofibromatosis. Am J Hum Genet 92(6):1001–1007
Betsholtz C, Keller A (2014) PDGF, pericytes and the pathogenesis of IBGC. Brain Pathol 24(4):387–395
Nicolas G, Jacquin A, Thauvin-Robinet C, Rovelet-Lecrux A, Rouaud O, Pottier C, Aubriot-Lorton MH, Rousseau S, Wallon D, Duvillard C, Béjot Y, Frébourg T, Giroud M, Campion D, Hannequin D (2014) A de novo nonsense PDGFB mutation causing idiopathic basal ganglia calcification with laryngeal dystonia. Eur J Hum Genet 22(10):1236–1238
Nicolas G, Rovelet-Lecrux A, Pottier C, Martinaud O, Wallon D, Vernier L, Landemore G, Chapon F, Prieto-Morin C, Tournier-Lasserve E, Frébourg T, Campion D, Hannequin D (2014) PDGFB partial deletion: a new, rare mechanism causing brain calcification with leukoencephalopathy. J Mol Neurosci 53(2):171–175
Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, Salem S, Radovanovic I, Grant AV, Adimi P, Mansouri N, Okada S, Bryant VL, Kong XF, Kreins A, Velez MM, Boisson B, Khalilzadeh S, Ozcelik U, Darazam IA, Schoggins JW, Rice CM, Al-Muhsen S, Behr M, Vogt G, Puel A, Bustamante J, Gros P, Huibregtse JM, Abel L, Boisson-Dupuis S, Casanova JL (2012) Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science 337(6102):1684–1688
Hertzog PJ, Williams BR (2013) Fine tuning type I interferon responses. Cytokine Growth Factor Rev 24(3):217–225
Yamada M, Masaki T, Mari T, Seiju K, Taguchi Y, Takashima S, Tanaka K, Touge T, Hatsuta H, Murayama S, Hayashi Y, Kaneko M, Ishiura H, Mitsui J, Atsuta N, Sobue G, Shimozawa N, Inuzuka T, Tsuji S, Hozumi I (2014) Evaluation of SLC20A2 mutations that cause idiopathic basal ganglia calcification in Japan. Neurology 82(8):705–712
Kasuga K, Konno T, Saito K, Ishihara A, Nishizawa M, Ikeuchi T (2014) A Japanese family with idiopathic basal ganglia calcification with novel SLC20A2 mutation presenting with late-onset hallucination and delusion. J Neurol 261(1):242–244
Zhu M, Zhu X, Wan H, Hong D (2014) Familial IBGC caused by SLC20A2 mutation presenting as paroxysmal kinesigenic dyskinesia. Parkinsonism Relat Disord 20(3):353–354
Taglia I, Mignarri A, Olgiati S, Menci E, Petrocelli PL, Breedveld GJ, Scaglione C, Martinelli P, Federico A, Bonifati V, Dotti MT (2014) Primary familial brain calcification: genetic analysis and clinical spectrum. Mov Dis 29(13):1691–1695
Lemos R, Oliveira M, Oliveira J (2013) Reporting a new mutation at the SLC20A2 gene in familial idiopathic basal ganglia calcification. Eur J Neurol 20:e43–e44
Rubino E, Giorgio E, Gallone S, Pinessi L, Orsi L, Gentile S, Duca S, Brusco A (2014) Novel mutation of SLC20A2 in an Italian patient presenting with migraine. J Neurol 261(10):2019–2021
Brighina L, Saracchi E, Ferri F, Gagliardi M, Tarantino P, Morzenti S, Musarra M, Patassini M, Annesi G, Ferrarese C (2014) Fahr’s disease linked to a novel SLC20A2 gene mutation manifesting with dynamic aphasia. Neurodegener Dis 4(3):133–138
Hayashi T, Legati A, Nishikawa T, Coppola G (2014) First Japanese family with primary familial brain calcification due to a mutation in the PDGFB gene: an exome analysis study. Psychiatry Clin Neurosci 69(2):77–83
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Taglia, I., Bonifati, V., Mignarri, A. et al. Primary familial brain calcification: update on molecular genetics. Neurol Sci 36, 787–794 (2015). https://doi.org/10.1007/s10072-015-2110-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-015-2110-8