
Abstract
Objective
To evaluate the potential impact of consistent use of similar treatments over a long period; it is essential to investigate the potential correlation between genetic variations that influence the expression or function of pharmacological targets for reducing lipid levels and the risk of developing rheumatoid arthritis.
Methods
We used variants in the following genes to conduct Mendelian randomization analyses: HMGCR (encoding the target for statins), PCSK9 (encoding the target for PCSK9 inhibitors, such as evolocumab and alirocumab), and NPC1L1 (encoding the target for ezetimibe). Data from lipid genetics consortia (173,082 sample size) were used to weight variations according to their correlations with low-density lipoprotein cholesterol (LDL-C). In two large datasets (total n = 19,562 cases, 501,655 controls). We conducted a meta-analysis of Mendelian randomization estimates, weighted by LDL-C levels, on the regional differences in the risk of rheumatoid arthritis using data from two large databases.
Results
We approached SMR and IVW-MR analyses to examine the relationship between target gene expression (including HMGCR, PCSK9, and NPC1L1) and LDL-C levels mediated by these genes with RA. The IVW-MR analysis revealed no significant association between genetically predicted LDL-C concentration and the risk of RA (OR = 0.88, 95% CI = 0.59–1.29; OR = 0.91, 95% CI = 0.67–1.23; OR = 0.81, 95% CI = 0.49–1.36; all p > 0.05). Similarly, our findings from the SMR approach provided no evidence to suggest that gene expression of HMGCR, PCSK9, and NPC1L1 was associated with the risk of RA (OR = 0.91, 95% CI = 0.79–1.05, p = 0.207; OR = 0.96, 95% CI = 0.85–1.09, p = 0.493).
Conclusions
Our results do not provide evidence to support the hypothesis that reducing LDL-C levels with statins, alirocumab, or ezetimibe effectively prevents the risk of developing RA. However, our study provides valuable insights into the assessment of lipid-lowering agents in RA, which can enhance our understanding of the condition and assist in clinical practice by aiding in the determination and monitoring of RA status to clinical response.
Key Points • Common lipid-lowering drugs such as statins, alirocumab, or evolocumab may not effectively prevent the development of rheumatoid arthritis. • This study provides clues for further exploration of the role of lipid-lowering therapy in other high-risk diseases, contributing to a deeper understanding of the potential effects of lipid-lowering treatment. |
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Rheumatoid arthritis (RA) is a persistent autoimmune disorder that predominantly impacts the hands and feet, leading to immune cell infiltration, synovial hyperplasia, pannus formation, and degradation of joint cartilage and bone [1, 2]. Although the precise etiology of RA remains unclear, it is widely acknowledged that a combination of genetic and environmental factors contributes to its onset [3]. RA affects a significant percentage of the global population with women being at higher risk than men [4]. Individuals with RA commonly encounter morning stiffness, which, if not addressed, can result in joint damage, deformities, and impaired functionality [5].
Statins, also known as HMG-CoA reductase inhibitors, were initially developed to treat lipid disorders and have been extensively studied for their effectiveness in reducing the incidence and mortality of cardiovascular diseases in primary and secondary prevention settings. While the beneficial effects of statins in reducing cardiovascular morbidity and mortality are well-established, their potential benefits in other conditions such as osteoporotic fractures and rheumatoid arthritis (RA) are still under investigation and have not yet been conclusively confirmed [6,7,8,9]. Therefore, further research is needed to validate their potential benefits in conditions of rheumatoid arthritis (RA). Patients with RA should undergo lipid testing due to their increased cardiovascular risk. Treatment decisions regarding statins should be personalized and made in collaboration with healthcare professionals.
The naturally occurring variations in genes that encode drug targets for lowering cholesterol can be used for Mendelian randomization (MR) to examine the therapeutic inhibition’s impact on disease outcomes, with less prone to confounding issues [10, 11]. Furthermore, MR analysis enables the investigation of the impact of drug targets on RA risk in the long term. This approach can simulate the pharmacological regulatory effects of drug targets in clinical trials and has been previously utilized to predict potential clinical benefits and adverse reactions of therapeutic interventions [12,13,14,15,16].
We used MR methods to examine the effects of drug targets for HMGCR (statin target), PCSK9 (PCSK9 inhibitor targets such as evolocumab and alirocumab), and NPC1L1 (ezetimibe target) on overall and subtype-specific risks of RA related to lowering cholesterol.
Materials and methods
Study design
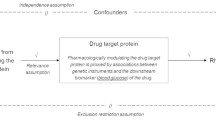
This two-sample MR study was based on summary-level data from publicly available genome-wide association studies (GWASs) and expression quantitative trait locus (eQTLs) studies (Fig. 1). All participants in these studies provided informed consent, and the studies received approval from their respective institutional review boards.

Overview of the design and methods used in this Mendelian
Selection of genetic instruments
This study investigated the effects of three lipid-lowering medications approved by the FDA, namely HMGCR inhibitors, PCSK9 inhibitors, and NPC1L1 inhibitors, on the risk of developing RA. The eQTLs of drug target genes were used as a proxy for exposure to each medication, with data collected from GTEx Consortium V8 or eQTLGen Consortium (Supplementary Table S1). Cis-eQTLs, defined as eQTLs within 1 Mb on either side of the encoded gene, were used to create genetic instruments [17,18,19]. Additionally, single nucleotide polymorphisms (SNPs) associated with LDL cholesterol levels at a genome-wide significance level (p < 5.0 × 10−8) within 100 kb windows from each drug’s target gene were chosen to validate the observed association using eQTLs as an instrument. The SNPs used in this study were identified from GWAS summary data of low-density lipoprotein (LDL) cholesterol levels obtained from the Global Lipids Genetics Consortium, which included 173,082 individuals. Only common SNPs with a minor allele frequency (MAF) > 1% were included in the analysis. All participants in the studies providing the data gave informed consent, and the studies received approval from the relevant institutional review boards [20]. To enhance the effectiveness of the instruments for each drug, a total of 22 SNPs were selected from three genes (HMGCR, PCSK9, and NPC1L1), with seven SNPs chosen for HMGCR inhibitors, 12 SNPs for PCSK9 inhibitors, and three SNPs for NPC1L1 inhibitors. These SNPs were specifically chosen to avoid significant linkage disequilibrium with one another, ensuring that the instrument power remains high while minimizing any potential biases.
Instruments selection and RA outcomes
From the eQTLGen and GTEx Consortium data, we discovered a total of 921 cis-eQTLs for the gene HMGCR, 24 cis-eQTLs for PCSK9, and 11 cis-eQTLs for NPC1L1, which are the target genes of the respective drugs. For each target gene, we selected the most significant cis-eQTL SNP as the genetic instrument for our analysis. From the Global Lipids Genetics Consortium GWAS summary data on LDL cholesterol levels, we selected 7, 12, and 3 SNPs within or near the genes HMGCR, PCSK9, and NPC1L1, respectively, to serve as instrumental variables (Table 1). All of the instruments had F-statistics greater than 30, reducing the potential for weak instrument bias in our analyses (Supplementary Table S3).
The summary statistics for rheumatoid arthritis (RA) used in this study were obtained from a meta-analysis of genome-wide association studies (GWAS), involving 14,361 cases and 43,923 controls of European ancestry [21]. Additionally, for the replication cohort, summary statistics for RA were acquired from a large-scale meta-analysis conducted by Eunji Ha et al. [22]. The study included three large case-control sets of East Asian and European populations. To eliminate population stratification bias, we specifically utilized data from European populations comprising 5201 cases and 457,732 controls and retrieved SNPs and their accompanying summary data from studies that exclusively included populations of European ancestry. The clinical diagnosis of RA was established according to the criteria of the ICD-10.
Positive control analyses
To validate our research and ensure the robustness of our modeling strategy, we applied the same method to predict the long-term effects of modulating these targets on cardiometabolic outcomes. As part of a positive control analysis, we utilized the LDL‐C summary statistics along with genetic data on the risk of coronary artery disease (CAD). These diseases are known to be influenced by LDL‐C modification.
Our positive control analysis incorporated a meta-analysis of GWAS on CAD (60,801 cases and 123,504 controls) [23]. It is important to note that these analyses were limited to individuals of European ancestry. The associations we observed between genetic predictions of exposure to lipid-lowering drug classes and a lower risk of CAD in this positive control analysis provide further support for the effectiveness of our approach.
Statistical analyses
Primary MR analysis
To estimate the effect of eQTL, we used the IVW-MR and SMR methods based on the pooled data. To combine the effect estimates obtained from genetic variations related to LDL cholesterol levels, we used the inverse-variance-weighted MR (IVW-MR) approach.
For evaluating the relationship between the expression of HMGCR, PCSK9, and NPC1L1 and the prognosis of rheumatoid arthritis (RA), we utilized the SMR method. A total of 921 SNPs for HMGCR, 24 SNPs for PCSK9, and 11 SNPs for NPC1L1 were obtained from eQTLGen and GTEx datasets. We then conducted SMR analyses (https://cnsgenomics.com/software/smr/#Overview) using the most significant cis-eQTL SNPs as instrumental variables for the target genes of each lipid-lowering medication. The results of these analyses are presented in Supplementary Table S7 [24].
The correlation between HMGCR, PCSK9, and NPC1L1 expressions and RA prognosis was evaluated by the SMR method. The cis-eQTL results yielded 921, 24, 11, and 161 SNPS of the drug target genes HMGCR, PCSK9, and NPC1L1 from eQTLGen and GTEx, respectively. Next, to conduct the SMR analysis, we utilized the SMR software version 1.03, which enabled allele harmonization and effect estimation. The most significant cis-eQTL SNPs (rs6453133, rs472495, rs41279633, and rs4665179) were used as IVs for each lipid-lowering drug target gene, as indicated in Supplementary Table S7. For allele harmonization and analysis, we employed R software version 4.1.0 along with the TwoSampleMR package.
Sensitivity analysis
To ensure the strength of the IVs, we assessed their instrument strength using the F-statistic. Only SNPs with an F-statistic greater than 10 were included to minimize bias from weak instruments [25]. To validate the IVs, we conducted positive control studies. For the eQTL instruments, we examined the correlation between the exposures of interest and LDL cholesterol levels since reducing LDL cholesterol is a well-established effect of lipid-lowering drugs. For the LDL cholesterol GWAS instrument, we assessed the association between relevant exposures and coronary heart disease, which is the primary indication for lipid-lowering medications. To determine if the observed association between gene expression and the RA in the SMR analysis was due to linkage rather than true causation, we performed the HEIDI test [26]. A HEIDI test result with a p-value less than 0.01 suggests the association might be due to linkage [27]. To assess the possibility of horizontal pleiotropy, we conducted SMR analysis on additional nearby genes within a 1 Mb window whose expression was significantly associated with the instrumental variant. This helped evaluate if the expression of these genes was linked to the outcome of RA and assess the risk of horizontal pleiotropy. For assessing heterogeneity, we employed the Cochran Q test in the IVW-MR approach, where p-values less than 0.05 indicate evidence of heterogeneity [28]. To detect horizontal pleiotropy in the instrumental variables, we used MR-PRESSO analysis. The presence of directed horizontal pleiotropy was examined using the intercept term in MR-Egger regression, with a p-value less than 0.05 suggesting such evidence [29]. To detect horizontal pleiotropy in the instrumental variables, we used MR-PRESSO analysis. The presence of directed horizontal pleiotropy was examined using the intercept term in MR-Egger regression, with a p-value less than 0.05 suggesting such evidence [30]. To account for significant correlations in a multivariable MR analysis, we utilized R software version 4.1.0. To adjust for multiple testing, the Bonferroni correction was applied, considering p < 0.05 as strong evidence (three exposures and one outcome) and 0.017 < p < 0.05 as suggestive evidence (Fig. 2).

IVW-MR association between LDL cholesterol mediated by gene HMGCR, PCSK9 or NPC1L1 and RA outcomes
Results genetic
Causal effect of LDL-C level mediated by target genes on RA via MR analyses
In the discovery cohort, the IVW-MR analysis revealed no significant causal effect of HMGCR, PCSK9, and NPC1L1-mediated LDL-C levels on the risk of RA (OR = 0.75, 95% CI = 0.566–0.998; OR = 0.90, 95% CI = 0.67–1.24; OR = 0.68, 95% CI = 0.30–1.55). Further validation was conducted using data from the UKB consortium, followed by a meta-analysis of the discovery and validation cohorts. The meta-analysis results also indicated no significant causal effect of HMGCR, PCSK9, and NPC1L1-mediated LDL-C levels on the risk of RA (OR = 0.995, 95% CI = 0.991–0.999; OR = 0.999, 95% CI = 0.996–1.002; OR = 0.998, 95% CI = 0.991–1.006). Overall, our findings consistently suggest that there is no substantial causal relationship between LDL-C levels mediated by HMGCR, PCSK9, and NPC1L1 and the risk of RA (OR = 0.88, 95% CI = 0.59–1.29, p = 0.092; OR = 0.91, 95% CI = 0.67–1.23, p = 0.537; OR = 0.81, 95% CI = 0.49–1.36, p = 0.502) (Supplementary Tables S2, S3, and S4).
Additionally, the Cochran Q test in the IVW-MR analysis did not find any evidence of heterogeneity for all reported results (all p > 0.05; Supplementary Tables S2 and S3). Furthermore, sensitivity analyses consistently demonstrated a lack of causal association (all p > 0.05). Additionally, the MR-PRESSO analysis revealed no significant evidence of heterogeneity (all p > 0.05). Both the intercept term in MR-Egger regression and MR-PRESSO analysis indicated no significant overall horizontal pleiotropy (all p > 0.05; Supplementary Tables S2 and S3). By conducting these rigorous tests, we have established the robustness of our findings and minimized the influence of confounding factors that could affect the causal relationship under investigation (Fig. 3).

SMR association between expression of gene HMGCR, PCSK9, or NPC1L1 and RA outcomes
SMR analyses
To ensure the validity of our findings, we conducted several tests in this study. For SMR analysis, the HEIDI test indicated that the observed associations were not influenced by linkage (p > 0.01) (Supplementary Tables S5 and S6). The SMR method was used to evaluate the correlation between HMGCR, PCSK9, and NPC1L1 expressions and RA outcome. In our study, we obtained a total of 921, 24, 11, and 161 SNPs for cis-eQTL results on the drug target genes HMGCR, PCSK9, and NPC1L1 from eQTLGen or GTEx consortium, respectively. We then performed SMR analysis using the most significant cis-eQTL SNPs (rs6453133, rs472495, and rs41279633) as IVs for each lipid-lowering drug target gene (Supplementary Tables S5 and S6). However, we did not find any evidence of a significant association between HMGCR and PCSK9 gene expression and the risk of RA (OR = 0.91, 95% CI = 0.79–1.05, p = 0.207), PCSK9 (OR = 0.96, 95% CI = 0.85–1.09, p = 0.493).
Positive control analysis
To further validate our research, we performed a positive control study to predict the effect of long-term modulation of these drug targets on cardiometabolic outcomes. We applied the same method and found that genetic predictions of exposure to all lipid-lowering drug classes were associated with a lower risk of CAD. Additionally, we observed significant associations between exposure to each drug and LDL cholesterol levels when using the proposed eQTLs as instrumental variables. Furthermore, using LDL cholesterol GWAS-proposed instruments, we found significant associations between exposure to each drug and coronary heart disease. These findings provide further confirmation of the efficacy of the selected genetic instruments and support the validity of our research (Supplementary Tables S7 and S8).
Discussion
In our study, we adopted an integrated approach that included conventional IVW-MR and SMR analyses to investigate the causal effects of genetically determined lipid-related traits and lipid-lowering medications on the risk of RA in a European population. Our findings do not provide genetic support for the use of statins, alirocumab, or ezetimibe as treatments for the prevention of RA. These results suggest that these lipid-lowering medications may not have a significant causal effect on RA risk in the population studied.
Some studies have indicated that statins, known for their inhibitory effects on bone morphogenetic protein and osteoclastogenesis, may have potential bone-protective properties in RA [31, 32]. In an experimental rat model of arthritis, female Lewis rats were given simvastatin either 4 days before arthritis induction or 8 days after induction. Simvastatin was found to prevent early and late-stage joint inflammation, potentially by reducing the influx of macrophages into the joints [33]. Furthermore, simvastatin inhibited late-stage periarticular bone destruction and preserved periarticular bone density. These findings indicate that statin medications might protect the periarticular bone within RA joints by suppressing inflammation-induced bone resorption.
Clinical research has shown promising results in the use of statin medications for the treatment of RA. For example, a small-scale clinical study demonstrated a significant reduction in disease activity in RA patients treated with simvastatin [34]. Another trial with atorvastatin showed a moderate improvement in disease activity, reduced inflammatory markers, and improved lipid profiles in RA patients [35]. In line with these findings, David McCarey and colleagues reported in their randomized controlled trial on the use of atorvastatin in patients with RA that the treatment group showed significant reductions in inflammatory markers, disease activity, total cholesterol, and triglyceride levels. However, they concluded that further larger-scale studies are needed to explore the potential of statin drugs in RA. We agree with their conclusion but emphasize that these studies should have sufficient power to examine hard endpoints such as mortality and major cardiovascular events [36]. However, they noted the need for further larger-scale studies to explore the full potential of statin drugs in RA treatment. Therefore, larger-scale studies with longer follow-up periods are needed to confirm the efficacy and optimal dosage of statin medications in preventing bone destruction in RA. These future studies will provide more robust evidence regarding the potential benefits of statins in managing RA and its associated complications. Additionally, these studies must examine hard endpoints such as mortality and major cardiovascular events to provide a comprehensive evaluation of the benefits and risks associated with statin use in RA.
Our IVW-MR and SMR analyses yielded consistent results, indicating no association between lipid-related factors and the risk of developing RA in individuals of European ancestry. These findings suggest that the previously observed contradictory results may be attributed to unmeasured or poorly measured confounding factors in observational studies, which can introduce confounding and reverse causality. Furthermore, it is important to consider that the relationship between lipid levels and the risk of RA may be influenced by the presence of cerebrovascular disease. This suggests that the impact of lipid levels on RA risk may be directly mediated by the presence or development of cerebrovascular disease. Understanding this potential mediating factor is crucial in comprehensively evaluating the association between lipid levels and RA risk and may have implications for the management and treatment of RA patients with concurrent cerebrovascular disease. Further research is needed to investigate and elucidate the mechanisms underlying this relationship.
It is worth noting that lipid bioactive products have been proposed to play a significant role in rheumatoid arthritis (RA). Lipid levels can fluctuate with changes in RA disease activity and treatment. In a subgroup analysis of early aggressive rheumatoid arthritis treatment, all treatment groups, including DMARDs MTX monotherapy, MTX, SSZ, HCQ triple therapy, or MTX + etanercept treatment, showed an average increase in LDL cholesterol levels by 6 mg/dl after 30 months [37]. This finding emphasizes the importance of closely monitoring cholesterol levels in RA patients, not only due to the increased risk of coronary artery disease but also due to the lipid changes associated with RA treatment. According to EULAR guidelines, healthcare professionals recommend regular lipid profile checks for RA patients when they achieve low disease activity after initiating RA treatment. This monitoring helps ensure early detection and appropriate management of lipid-related abnormalities in RA patients.
Interestingly, addressing modifiable risk factors such as dyslipidemia and tightly controlling RA is crucial for minimizing overall CAD risk. Statin medications are highly effective in reducing CAD risk in this population, and numerous studies support their use [38, 39]. The cholesterol treatment guidelines from the European Society of Cardiology (ESC) and the American College of Cardiology/American Heart Association recommend lifestyle modifications and the addition of statin therapy based on the 10-year high-risk for CAD events (fatal or non-fatal coronary heart disease or stroke) and LDL cholesterol levels [40]. By closely monitoring cholesterol levels and addressing modifiable risk factors, healthcare professionals aim to mitigate the risk of CAD and ensure optimal management of RA in patients, as recommended by the EULAR guidelines.
Our study has several notable strengths that set it apart from previous research. Firstly, we conducted both IVW-MR and SMR analyses to assess the causal effects of genetically determined lipid-related traits and lipid-lowering medications on the risk of GWAS and eQTL. This comprehensive approach allowed us to examine these relationships from multiple perspectives and provide a more robust analysis. Furthermore, the SMR analysis framework was particularly valuable in our study as it enabled us to investigate pleiotropic associations and potential causal relationships between gene expression levels and RA. By utilizing summary-level data from GWAS and eQTL studies, we were able to reduce biases associated with reverse causality and confounding factors, strengthening the validity of our findings. Lastly, our study’s results are less susceptible to population stratification bias due to our deliberate restriction of the aggregated data to individuals of European ancestry. This decision helped ensure a more homogeneous study population, enhancing the reliability and generalizability of our conclusions.
Although our study is innovative, it is important to acknowledge several limitations. Firstly, the sample size of RA cases in our study was relatively small, which may have limited the statistical power of our results. Secondly, the use of summary-level data instead of individual-level data may have restricted our ability to perform stratified analyses or adjust for additional confounding factors. Thirdly, while our study provides valuable insights into the relationship between gene expression and rheumatoid arthritis outcomes based on GWAS data from European and East Asian populations, it is important to note that the generalizability of our findings to other ancestral groups, such as African-Americans, Caucasians, Hispanics, and other populations, may be limited. The potential differences in genetic architecture and environmental factors across diverse ancestral groups highlight the need for future studies to encompass a broader range of ancestries to ensure the broader applicability of our results. Additionally, the unavailability of specific lipid markers or parameter data from publicly accessible GWAS in European populations limited our exploration of causality about these markers. Lastly, while the liver is a key organ in lipid metabolism, we did not have access to liver tissue-specific eQTL data, which could have provided additional insights. Future research should aim to address these limitations, including incorporating liver eQTL data, to improve our understanding of the relationship between lipid-related traits and RA risk.
Conclusions
Our findings do not provide support for the hypothesis that lowering LDL-C levels with statins, alirocumab, or ezetimibe targets is an effective preventive strategy for RA risk. However, it is important to note that further confirmation is needed. To establish conclusive evidence, prospective and large-scale clinical trials are required. These trials would provide more robust data to determine the potential benefits or lack thereof in using these interventions for the prevention of RA.
Data availability
The original contributions presented in the study are included in the article/supplementary material; further inquiries can be directed to the corresponding author/s.
References
Smith MH, Berman JR (2022) What Is rheumatoid arthritis? JAMA 327(12):1194. https://doi.org/10.1001/jama.2022.0786
Szekanecz Z, Koch AE, Tak PP (2011) Chemokine and chemokine receptor blockade in arthritis, a prototype of immune-mediated inflammatory diseases. Neth J Med 69(9):356–66
Firestein GS (2003) Evolving concepts of rheumatoid arthritis. Nature 423(6937):356–361. https://doi.org/10.1038/nature01661
Ngo ST, Steyn FJ, McCombe PA (2014) Gender differences in autoimmune disease. Front Neuroendocrinol 35(3):347–369. https://doi.org/10.1016/j.yfrne.2014.04.004
Turesson C, O’Fallon WM, Crowson CS et al (2003) Extra-articular disease manifestations in rheumatoid arthritis: incidence trends and risk factors over 46 years. Ann Rheum Dis 62(8):722–727. https://doi.org/10.1136/ard.62.8.722
Nurmohamed MT, Dijkmans BA (2009) Dyslipidaemia, statins and rheumatoid arthritis. Ann Rheum Dis 68(4):453–455. https://doi.org/10.1136/ard.2008.104497
Heslinga M, Nurmohamed MT (2016) Cardiovascular Disease reduction in rheumatoid arthritis by statins: the final evidence? J Rheumatol 43(11):1950–1952. https://doi.org/10.3899/jrheum.161131
Koch CA, Krabbe S, Hehmke B (2018) Statins, metformin, proprotein-convertase-subtilisin-kexin type-9 (PCSK9) inhibitors and sex hormones: immunomodulatory properties? Rev Endocr Metab Disord 19(4):363–395. https://doi.org/10.1007/s11154-018-9478-8
Semb AG, Holme I, Kvien TK et al (2011) Intensive lipid lowering in patients with rheumatoid arthritis and previous myocardial infarction: an explorative analysis from the incremental decrease in endpoints through aggressive lipid lowering (IDEAL) trial. Rheumatology 50(2):324–329. https://doi.org/10.1093/rheumatology/keq295. (Oxford)
Smith GD, Ebrahim S (2003) “Mendelian randomization”: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 32(1):1–22. https://doi.org/10.1093/ije/dyg070
Yarmolinsky J, Wade KH, Richmond RC et al (2018) Causal inference in cancer epidemiology: what is the role of Mendelian randomization? Cancer Epidemiol Biomarkers Prev 27(9):995–1010. https://doi.org/10.1158/1055-9965.EPI-17-1177
Zhao YJ, Ong J, Goadsby PJ (2020) Emerging treatment options for migraine. Ann Acad Med Singap 49(4):226–235
Ference BA, Majeed F, Penumetcha R et al (2015) Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both: a 2 x 2 factorial Mendelian randomization study. J Am Coll Cardiol 65(15):1552–1561. https://doi.org/10.1016/j.jacc.2015.02.020
Ference BA, Robinson JG, Brook RD et al (2016) Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. N Engl J Med 375(22):2144–2153. https://doi.org/10.1056/NEJMoa1604304
Stroup DF, Berlin JA, Morton SC et al (2000) Meta-analysis of observational studies in epidemiology: a proposal for reporting. Meta-analysis Of Observational Studies in Epidemiology (MOOSE) group. JAMA 283(15):2008–12. https://doi.org/10.1001/jama.283.15.2008
Plenge RM, Scolnick EM, Altshuler D (2013) Validating therapeutic targets through human genetics. Nat Rev Drug Discov 12(8):581–594. https://doi.org/10.1038/nrd4051
Li Z, Zhang B, Liu Q et al (2023) Genetic association of lipids and lipid-lowering drug target genes with non-alcoholic fatty liver disease. EBioMedicine 90:104543. https://doi.org/10.1016/j.ebiom.2023.104543
Li Z, Zhang B, Liu Q et al (2023) Genetic association of lipids and lipid-lowering drug target genes with non-alcoholic fatty liver disease. EBioMedicine 90:104543. https://doi.org/10.1016/j.ebiom.2023.104543
Williams DM, Finan C, Schmidt AF et al (2020) Lipid lowering and Alzheimer disease risk: a Mendelian randomization study. Ann Neurol 87(1):30–39. https://doi.org/10.1002/ana.25642
Willer CJ, Schmidt EM, Sengupta S et al (2013) Discovery and refinement of loci associated with lipid levels. Nat Genet 45(11):1274–1283. https://doi.org/10.1038/ng.2797
Okada Y, Wu D, Trynka G et al (2014) Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506(7488):376–381. https://doi.org/10.1038/nature12873
Ha E, Bae SC, Kim K (2021) Large-scale meta-analysis across East Asian and European populations updated genetic architecture and variant-driven biology of rheumatoid arthritis, identifying 11 novel susceptibility loci. Ann Rheum Dis 80(5):558–565. https://doi.org/10.1136/annrheumdis-2020-219065
Nikpay M, Goel A, Won HH et al (2015) A comprehensive 1,000 genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet 47(10):1121–1130. https://doi.org/10.1038/ng.3396
Zhu Z, Zhang F, Hu H et al (2016) Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 48(5):481–487. https://doi.org/10.1038/ng.3538
Burgess S, Thompson SG (2011) Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol 40(3):755–764. https://doi.org/10.1093/ije/dyr036
Zhu Z, Zhang F, Hu H et al (2016) Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 48(5):481–487. https://doi.org/10.1038/ng.3538
Chauquet S, Zhu Z, O’Donovan MC et al (2021) Association of antihypertensive drug target genes with psychiatric disorders: a Mendelian randomization study. JAMA Psychiat 78(6):623–631. https://doi.org/10.1001/jamapsychiatry.2021.0005
Higgins JP, Thompson SG, Deeks JJ et al (2003) Measuring inconsistency in meta-analyses. BMJ 327(7414):557–560. https://doi.org/10.1136/bmj.327.7414.557
Burgess S, Thompson SG (2017) Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol 32(5):377–389. https://doi.org/10.1007/s10654-017-0255-x
Verbanck M, Chen CY, Neale B et al (2018) Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet 50(5):693–698. https://doi.org/10.1038/s41588-018-0099-7
Nurmohamed MT, Dijkmans BA (2009) Dyslipidaemia, statins and rheumatoid arthritis. Ann Rheum Dis 68(4):453–455. https://doi.org/10.1136/ard.2008.104497
Wald NJ, Law MR (2003) A strategy to reduce cardiovascular disease by more than 80%. BMJ 326(7404):1419. https://doi.org/10.1136/bmj.326.7404.1419
Funk JL, Chen J, Downey KJ et al (2008) Bone protective effect of simvastatin in experimental arthritis. J Rheumatol 35(6):1083–1091
Abud-Mendoza C, de la Fuente H, Cuevas-Orta E et al (2003) Therapy with statins in patients with refractory rheumatic diseases: a preliminary study. Lupus 12(8):607–611. https://doi.org/10.1191/0961203303lu429oa
McCarey DW, McInnes IB, Madhok R et al (2004) Trial of Atorvastatin in Rheumatoid Arthritis (TARA): double-blind, randomised placebo-controlled trial. Lancet 363(9426):2015–2021. https://doi.org/10.1016/S0140-6736(04)16449-0
McCarey DW, McInnes IB, Madhok R et al (2004) Trial of Atorvastatin in Rheumatoid Arthritis (TARA): double-blind, randomised placebo-controlled trial. Lancet 363(9426):2015–2021. https://doi.org/10.1016/S0140-6736(04)16449-0
Navarro-Millan I, Charles-Schoeman C, Yang S et al (2013) Changes in lipoproteins associated with methotrexate or combination therapy in early rheumatoid arthritis: results from the treatment of early rheumatoid arthritis trial. Arthritis Rheum 65(6):1430–1438. https://doi.org/10.1002/art.37916
An J, Alemao E, Reynolds K et al (2016) Cardiovascular outcomes associated with lowering low-density lipoprotein cholesterol in rheumatoid arthritis and matched nonrheumatoid arthritis. J Rheumatol 43(11):1989–1996. https://doi.org/10.3899/jrheum.160110
DeBose-Boyd RA (2018) Significance and regulation of lipid metabolism. Semin Cell Dev Biol 81:97. https://doi.org/10.1016/j.semcdb.2017.12.003
Crowson CS, Rollefstad S, Ikdahl E et al (2018) Impact of risk factors associated with cardiovascular outcomes in patients with rheumatoid arthritis. Ann Rheum Dis 77(1):48–54. https://doi.org/10.1136/annrheumdis-2017-211735
Acknowledgements
We want to acknowledge the International Headache Genetics Consortium for providing summary data on migraine. We want to acknowledge the participants and investigators of the FinnGen study and the UK Biobank. We also want to acknowledge the participants and investigators of all other studies.
Funding
This work was supported by the Shanghai grassroots famous old Chinese medicine experts inheritance studio construction project (2020JCGZS-018) and Shanghai Xuhui District Medical Research Project (SHXH202221).
Author information
Authors and Affiliations
Contributions
LQ developed the protocol, participated in the literature search, extracted data, and drafted the manuscript; SL and KM were responsible for the analysis and interpretation of the data; J.Y was responsible for the critical revision of the manuscript for important intellectual content. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Ethics approval
Since we utilized publicly available GWAS summary data or published studies, ethical committee approval was not required for this manuscript.
Informed consent
Not applicable.
Disclosures
None.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Qiao, L., Lv, S., Meng, K. et al. Genetically proxied therapeutic inhibition of lipid-lowering drug targets and risk of rheumatoid arthritis disease: a Mendelian randomization study. Clin Rheumatol 43, 939–947 (2024). https://doi.org/10.1007/s10067-023-06837-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-023-06837-9