
Abstract
Gout is a disease with acute and/or chronic inflammation and tissue damage induced by the precipitation of monosodium urate crystal (MSU) crystals in bone joints, kidneys, and subcutaneous sites. In recent years, with the continuous research on gout animal models and patient clinical investigations, the mechanism of inflammation activation of gout has been further discovered. Studies have shown that pro-inflammatory factors such as interleukin (IL)-1β, IL-8 and IL-17, NLRP3 inflammasome, and tumor necrosis factor alpha (TNF-α), anti-inflammatory factors such as IL-10, IL-37 are all involved in the MSU-induced gout inflammatory process. And the immune cells in gout, including neutrophils, monocytes/macrophages, and lymphocytes, all play important roles in the pathogenesis of gout. In this review, we mainly emphasize the understanding of various cytokines, inflammasome, and immune cells involved in the onset of gout, in order to provide a systematic and theoretical basis for the novel exploration of inflammatory therapeutic targets for gout.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
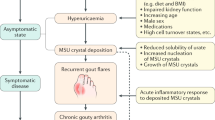
Gout is a crystal-associated joint disease caused by the precipitation of MSU, which is directly related to hyperuricemia caused by a disorder of purine metabolism and/or a decrease in uric acid excretion. The development of gout can be divided into several stages. Hyperuricemia is a prerequisite for the development of gout, and a significant concentration-dependent relationship exists between serum urate concentration and gout [1]. However, most of the patients with hyperuricemia do not have gout, suggesting that other stages in the clinical development of gout are also important since the clinical deposition of MSU crystals [2] (Fig. 1). Great changes have taken place in the current human living environment, lifestyle, and dietary structure, concurrent with the incidence of gout increasing year by year. More and more researchers are studying the pathogenesis of gout in terms of inflammatory factors and immunology. Through the participation of macrophages, monocytes and neutrophils, TNF-α, some interleukins (IL-1β, IL-8, IL-10, IL-17, IL-37), and NLRP3 inflammasome mediate the development of gout, inducing a series of cascade inflammatory amplification reactions [3]. This article briefly reviews the research progress of inflammatory mediators in MSU-induced gout inflammatory response, to further understand the pathogenesis of gout inflammation and to provide systematic theoretical basis and ideas for targeted treatment and prevention of this immune inflammatory disease.

The mechanism of hyperuricemia. Hyperuricemia is caused by the overproduction and underexcretion of urate. Underexcretion of urate occurs mainly in the proximal tubules of the kidneys, but the gut is also involved in the development of hyperuricemia. Many transmembrane transporters are very important for urate reuptake and secretion in the kidney, including ABC transporter G family member 2 (ABCG2), glucose transporter type 9 (GLUT9), and organic anion transporters (OATs). In the gut, mutations in the ABCG2 gene can reduce the function of the ABCG2 transporter, thereby hindering the excretion of urate. When MSU crystals are engulfed by monocytes, the NLRP3 inflammasome are further activated by Toll-like receptors TLR2 and TLR4, thereby pro-inflammatory IL-1β produced, and mature IL-1β formed after cleaved by active caspase-1
TLR recognition mode of monosodium urate crystal
The traditional recognition mechanism mainly refers to the interaction between the plasma membrane protein receptor and the ligand to trigger the downstream signaling pathway. MSU crystal itself does not contain any protein structure and cannot activate immune cells and tissue cells by traditional recognition mechanisms. The surface of MSU crystal is very hydrophilic and can adsorb regulatory proteins and other serum factors. IgG can adhere to the surface of MSU crystals and is related to the initiation of inflammation. One early study suggested that the phagocytosis of MSU crystals by macrophages requires the soluble CD14 coated on them [4], because CD14 is a pattern recognition molecule expressed by phagocytes and can interact with Toll-like receptors (TLRs) to mediate immune inflammatory responses. After macrophages engulf MSU crystals, they can cause a large amount of cathepsin B release and activate the NLRP3 inflammasome. Interestingly, researches had shown that the inflammatory potential of MSU crystals varied according to proteins coated on the crystal surfaces, and that the coated proteins changed during inflammation states [5]. Coating of the crystals by different proteins may modify their inflammatory potential, for example, IgG coating MSU crystals may enhance the inflammation; however, apolipoprotein B can displace the IgG by competitively coating sites on crystals and can contribute in part to the resolution of the acute gouty arthritis as the inflammation subside [6, 7]. Other studies showed that the inflammatory potential of MSU crystals increased with IgG coating while it decreased with ApoE or LDL coating [8, 9].
Uric acid is the end product of purine metabolism in the body, and MSU is one of the causes of gout. TLRs are important pattern recognition receptors (PRRs) in the innate immune system and are widely involved in the recognition and perception of pathogen-associated molecular patterns (PAMPs). As a kind of dangerous-associated molecular patterns (DAMPs), MSU crystal activates innate immunity mainly through a mechanism similar to PAMPs, resulting in a series of inflammatory factors-induced cascade amplification reactions [10]. TLR is a type I transmembrane receptor and mainly contains two important parts: one is the extracellular leucine-rich repeats which can recognize PAMPs and participate in the formation of TLRs dimers combined with PAPMs [11], another is the cytoplasmic Toll/IL-1 receptor (IL-1R) domain which is required for the downstream signaling pathway to activate nuclear transcription factor (NF-κB). NF-κB is a rapidly activated transcription factor of MSU crystals, which is mainly involved in MSU-induced cell activation and induces IL-1β production [12]. It has been found that the expression of IL-1β and TNF-α were decreased in mice lacking TLR2, TLR4, or MYD88 gene in the inflammatory responses induced by MSU in the animal model [13]. However, other studies suggested different views on the signaling pathways of MSU crystal-activated TLRs. Chen et al. [14] confirmed that the neutrophil influx was unaffected when MSU crystal stimulated the mice lacking TLR-1, TLR-2, TLR-3, TLR-4, TLR-6, TLR-7, TLR-9, and TLR-11, suggesting these TLRs may not be necessary in MyD88-dependent IL-1β activation, and other factors may be involved in this process. They also demonstrated the production of pro-inflammatory cytokines IL-1β, KC, and MIP-2 and neutrophil aggregation were reduced in MyD88-deficient mice stimulated with MSU crystals. Therefore, as a “danger signal” released by the injured cells or the dead cells, MSU crystal activates TLRs, next stimulates NF-kB through MyD88 signaling pathway, and then initiates the transcription and expression of inflammatory cytokines and chemokines, such as the transcription and expression of the pro-IL-1β gene, and ultimately leads to the production and release of IL-1β. These studies indicated that the inflammatory response induced by MSU crystals required a cellular adapter protein called MyD88, and the IL-1 signaling pathway played a central role in gout inflammation through the IL-1R-MyD88 pathway.
The role of NLRP3 inflammasome in MSU-induced joint inflammation
The NLRP3 inflammasome is the most typical member of the NOD-like receptors (NLRs) and is similar to TLRs. It includes NLRP3 protein, ASC, and caspase-1 protease. The NLRP3 protein molecule consists of three domains, and from the C-terminus to the N-terminus is the leucine-rich-repeat domain (LRR), the intermediate domain NACHT (the nucleotide binding oligomerization domain, NOD) and pyrin domain (PYD), respectively. ASC consists of a N-terminal PYD domain and a C-terminal CARD domain that can recruit and activate the precursor caspase-1 protease through the CARD domain [15] (Fig. 2). The three parts of NLRP3 inflammasome play an important role in the onset of acute inflammation of gout. LRR can mediate the recognition of different ligands to identify dangerous signals, while the CARD domain and PYD domain can interact with downstream signaling proteins. Inflammasome activation has two processes that require at least two signals to complete (Fig. 3): the initial startup step shows rapid activation of NF-kB by recognition of DAMP and PAMP, resulting in the synthesis of Pro-IL-1β and increase expression of NLRP3 protein; the second step means inflammasome assembly that oligomerization of NLRP3 and ASC promotes hydrolysis of pro-caspase-1 protease, produces active caspase-1 protease, and then caspase-1 can cut the precursor cytokines pro-IL-1β and pro-IL-18 into biologically active forms [16].

NLRP3 inflammasome activation. NLRP3 inflammasome is composed of three parts: NOD-like receptor proteins (including LRR, NACHT intermediate domain, and PYD domain), ASC, and pro-caspase-1; these elements constitute NLRP3 cleavage and activation of caspase-1, thereby cleaving the precursor pro-IL-1β into mature IL-1β

The initiation and activation process of the NLRP3 inflammasome. Activation of the NLRP3 requires at least two signals: Signal 1 is mediated by microbial ligands recognized by TLRs or cytokines, and upregulates precursor IL-1β and NLRP3 protein levels by activating the NF-κB pathway. Signal 2 promotes the assembly of ASC and pro-caspase-1 which is mediated by a large number of PAMP or DAMP stimulation, then leads to the activation of the NLRP3. These include K+ efflux, Ca2+ influx, Cl− efflux, lysosomal destruction, and mitochondrial active oxygen (mtROS) production. The formation of inflammasome activates pro-caspase-1, and further cleaves pro-IL-1β and pro-IL-18
Current research proposes that NLRP3 activation includes multiple upstream signals, because there are many types and different structures of stimuli that activate NLRP3 inflammasome. Most of them are not mutually exclusive, mainly including the following (Fig. 3): First, potassium (K+) efflux: MSU can affect intracellular K+ levels by activating intracellular ATP-P2X7R signaling pathway, and further activate NLRP3, so K+ outflow is an important upstream event for NLRP3 activation [17,18,19,20]. Second, chloride (Cl−) efflux: some studies have shown that Cl− channel blockers and extracellular high Cl− levels can inhibit the activation of NLRP3, suggesting Cl− efflux plays an important role in the activation of NLRP3 [21,22,23,24]. Third, calcium ion influx (Ca2+): the intracellular Ca2+ level can also affect the activation of NLRP3, there are studies indicating that the secretion of IL-1β decreases after adding the Ca2+ chelator BAPTA-AM. Fourth, lysosomal destruction: mononuclear macrophages form phagosomes after phagocytosing MSU, then the stability of the lysosomal membrane can be disrupted, resulting in cathepsin B exudation. NLRP3 activation can be inhibited in macrophages lacking cathepsin B. Fifth, mitochondrial damage and active oxygen: the activation of NLRP3 by ROS and mitochondrial damage is a hot topic, but the regulatory mechanism is unclear. Nevertheless, mitochondrial autophagy can clear damaged and dysfunctional mitochondria, so it is important in NLRP3 activation [25, 26]. Studies have found that antioxidants can effectively inhibit the production of IL-1β by NLRP3, which indicates the role of ROS in NLRP3 activation [27]. NADPH is driven to generate ROS after phagocytosing MSU, release thioredoxin-interacting protein (TXNIP) from the antioxidant protein thioredoxin (TRX) and bind to the LRR region, make the dissociation of NLRP3 protein inhibitory protein and then activate NLRP3 [28]. Sixth, the role of trans-Golgi: phosphatidylinositol 4-phosphate (ptdIns4p) on the decentralized trans-Golgi network (dTGN) can mediate NLRP3 activation, which is important for the oligomerization of downstream ASC and caspase-1 activation [29].
A study found the number of inflammatory cell exudation was reduced when MSU was injected subcutaneously or intraperitoneally in mice with NLRP3 protein deletion [14, 30]. Another study demonstrated that MSU-induced inflammatory responses were attenuated, and the cleavage and activation of casepase-1 and IL-1β release were also reduced in mice with lacking the LRR structure [31]. ASC is an important linker between NLRP3 protein and pro-casepase-1 protease in joint inflammation of acute gout. Martinon et al. [32] found that MSU and CPPD are both involved in the activation of NLRP3 inflammasome by caspase-1, resulting in massive activation of macrophages and secretion of active IL-1 and IL-18 in vitro. Moreover, their experiments have also shown that MSU-induced peritonitis was weakened in ASC-deficient mice; simultaneously, the production of IL-1β and neutrophil exudation was decreased. These studies all fully demonstrate the NLRP3 inflammasome signaling pathway is pivotal in MSU recognition and triggering inflammatory responses.
Tumor necrosis factor-α as a crucial mediator in gout
TNF-α is a multifunctional inflammatory cytokine, mainly produced by monocytes and macrophages, which can activate neutrophils and lymphocytes to promote the synthesis and release of other cytokines. Levels of TNF-α in synovial fluid are elevated in patients with gout; therefore, uric acid crystal deposition induces the release of TNF-α and the production of IL-1β. In addition, TNF-α is speculated to be associated with inflammatory arthritis, which promotes the activation and secretion of ATP-mediated caspase-1 without microbial stimulation [33].
Other studies showed that TNF-α pretreatment can induce the activation and release of NLRP3 inflammasome in adipose tissue [34]. Except for the cytokines that are dependent on caspase-1 activation, MSU and CPPD can also induce TNF-α release, suggesting the presence of other crystal activation pathways that are independent on inflammasomes. An experiment [35] has shown that TNF-α is a potent inducer of neutrophil-expressing pro-IL-1β mRNA. Neither MSU nor TNF-α alone is effective in inducing IL-1β secretion when stimulating neutrophils; however, the TNF-α-pretreated human neutrophils stimulated by MSU crystal can significantly increase IL-1β release and caspase-1 activation. The above suggests that TNF-α initiation is essential for MSU-mediated inflammasome activation in neutrophils, and uric acid-induced IL-1β release was triggered by the induction of inflammatory factor TNF-α. In addition, Amaral et al. [36] have found that few neutrophils were recruited into the joint cavity in TNF-α-/- mice and TNFR1/2-/- mice. All of these indicates that TNF-α is important in the MSU crystal-induced inflammation. Some studies report that tumor necrosis factor alpha antagonists (etanercept) have achieved good results in the treatment of refractory gout.
Many interleukin cytokines involving in gout attack
Interleukin-like cytokines are the members in the family of cytokines that play important roles in transmitting information, activating and regulating immune cells. They can be divided into pro-inflammatory cytokines and anti-inflammatory cytokines according to their roles in the inflammatory response. The onset of acute gout involves multiple inflammatory factors, mainly including IL-1β, IL-6, IL-8, IL-10, IL-17, IL-37, and TNF-α.
Pro-inflammatory cytokines
IL-1β
IL-1β called “endogenous pyrogen” is a potent pro-inflammatory cytokine to induce the expression of adhesion molecules in vascular endothelial cells and to activate macrophages is its main biochemical activity [37, 38]. As an endogenous risk signal, MSU can be recognized by PRRs in the cell membrane and cytoplasm [13], for example, it can be recognized by Toll-like receptors and NOD-like-receptors, activating TLRs and NLRP3 inflammasome to produce IL-1β (Fig. 3). IL-1β is originally produced by mononuclear macrophages, which synthesis and release are strictly controlled. Its maturation process has two stages: first, MSU interacts with MyD88-dependent TLR and MyD88-dependent IL-1 receptor on the cell surface to activate NF-κB and the transcription of IL-1β and NLRP3 target genes, then to form inactive pro-IL-1β; subsequently, MSU crystals activate intracellular NLRP3 inflammasome, and make casepase-1 protease cleave pro-IL-1β to form mature IL-1β and release extracellularly [10]. Mature IL-1β is considered to be one of the important inflammatory mediators of acute gouty arthritis, and is the initiating factor that regulates inflammation. A study has showed that IL-1α or IL-1β could synergize with IL-23 to induce secretion of IL-17A by T cells in mice stimulated or not stimulated by TCR [39]. However, IL-23-induced IL-17A secretion was absent in IL-1R-deficiency mice. IL-17 can promote the maturation of various inflammatory cells and the synergistic effects of multiple inflammatory factors. Anyway, IL-1β releases heavily after MSU crystal activates TLRs, eventually leading to a strong inflammatory response in gout patients [10]. At present, studies have shown that IL-1 receptor antagonist (anakinra) is effective in the treatment of refractory gout, but its safety and effectiveness need to be further verified by more detailed clinical trials [40].
IL-8
IL-8 belongs to the CXC subfamily in the chemotactic cytokine family, mainly produced by monocyte-macrophages and neutrophils. The major biological activity of IL-8 is to trend neutrophils and eosinophils as well as lymphocytes to the local inflammation areas to cause inflammations or allergic reactions. As a potent chemotactic and neutrophil-activating cytokine, IL-18 can not only activate neutrophils and basophils, but also dilate blood vessels and promote proliferation of vascular and granulocyte. In fact, IL-8 also increases the release of human monocyte inflammatory cytokines, including IL-1β, IL-6, and TNF-α, which can further modulate the inflammatory response. MSU stimulates synovial endothelial cells to significantly induce IL-8 mRNA production, thereby activating and trending neutrophil aggregation. Moreover, neutrophil-dependent inflammation is dependent on the induction of IL-8 expression by MSU crystals, suggesting that IL-8 release is closely related to the onset of acute gout inflammation [41]. MSU crystal stimulates monocytes and neutrophils in peripheral blood, leading to IL-8 production up to abnormal levels. IL-8 locally accumulates and continues to affect inflammatory response [42, 43].
IL-17
IL-17 is a pro-inflammatory cytokine produced primarily by activated CD4+CD45RO+ memory cells [41]. It is currently believed that IL-17 exerts its biological effects mainly through two pathways: the NF-κB-DNA pathway and the MAP kinase pathway. In the presence of NF-κB signaling, uric acid crystals stimulate dendritic cells and promote the release of cytokines associated with Th17 polarization. IL-17 also promotes T cell activation and stimulates epithelial cells, endothelial cells, and fibroblasts to produce a variety of cytokines such as IL-6, IL-8, granulocyte-macrophage stimulating factor, and cellular adhesion molecule-1 [44]. In addition, IL-17 also stimulates macrophages to produce IL-1β, TNF-α, and IL-6 [45]. Studies have indicated that IL-17 has a strong chemotactic effect on neutrophils mainly achieved by inducing the release of CXC chemokines and the increase of IL-8 synthesis, which can be enhanced by TNF-α. Furthermore, neutrophil generation was also reduced in vitro animal models knocking out the IL-17 gene. Moreover, IL-17 promotes the maturation and differentiation of a variety of cells, and create synergies of multiple cytokines to amplify the inflammatory response. Liu et al. have discovered that serum IL-17 level was increased after 8 h of acute gout attack [46]. The increase of serum IL-17 level in patients with early gout acute attack was mainly caused by the production of IL-17+γδT cells, and the rapid activation of IL-17+γδT cells was more consistent with the acute gout attack. Correspondingly, they also found that serum IL-17 level and the proportion of IL-17+γδT cells in lymphocytes were decreased with the reduction of disease symptoms. Therefore, the IL-17 releasing by IL-17+γδT cells can be considered a potential pathological mechanism of acute gout attack.
Anti-inflammatory cytokines
IL-10
It is known that IL-10 is secreted mainly by monocytes, macrophages, T and B lymphocytes, and is a multifunctional cytokine that regulates cell growth and differentiation, participates in inflammatory and immune responses, and is currently recognized as a suppressive factor for inflammation and immune [47]. As a potent anti-inflammatory cytokine, IL-10 can inhibit the production of IL-1α, IL-1β, IL-6, IL-12, IL-18, and TNF-α produced by activating mononuclear macrophages, neutrophils, and eosinophils. During the acute inflammation of gout, IL-10 can inhibit the transcription of the corresponding genes of pro-inflammatory cytokines such as TNF-α and IL-β, and block their synthesis. Meanwhile, IL-10 also affects the stability of mRNA of pro-inflammatory cytokines. Chen [48] found that compared with wild-type mice, the inflammation of collagen arthritis (CIA) was more severe in mice lacking IL-10 (IL-10-). IL-10 restrained the expression of IL-33 in macrophages, selectively destroyed the NF-kB signal activated by IL-33 and blocked the response of pro-inflammatory cytokines and chemokines to IL-33, thereby ameliorating the autoimmune arthritis. A study found that the level of IL-10 in the articular fluid of patients with gout was significantly higher than that in patients with osteoarthritis and normal people [49]. In addition, IL-10 treatment can reduce the secretion of IL-1β and TNF-α by peripheral monocytes and laminar monocytes in patients with inflammatory bowel disease [50]. The above suggested that the elevation of anti-inflammatory factor IL-l0 may be involved in the self-regulation of acute inflammatory immunity in gout patients.
IL-37
IL-37 belongs to the seventh factor of the IL-1 family and is an anti-inflammatory cytokine that has been widely studied in recent years. IL-37 is mainly expressed in skin, lymph nodes, thymus, bone marrow, NK cells, B cells, and testes. IL-37 is produced when activating macrophages inhibit pro-inflammatory cytokines, and its anti-inflammatory effect is to inhibit the TLR-induced expression of pro-inflammatory cytokines IL-1β, IL-16, and IFN-γ, thereby to attenuate the T cell-mediated inflammation by downregulation of dendritic cell activity [51, 52]. Nold et al. [53] discovered that the inflammatory response in the IL-37 transgenic mice (IL-37tg) was lighter than the wild control mice injected respectively by bacterial lipopolysaccharide (LPS), showing that IL-37 has a strong anti-inflammatory effect. It was confirmed that recombinant human IL-37 (rhIL-37) can significantly reduce the recruitment of neutrophils and monocytes, weaken the pathological joint inflammation by reducing the release of pro-inflammatory cytokines and chemokines [54] (Table 1). rhIL-37 can also inhibit the MSU-induced innate immune response by enhancing the expression of Smad3 and IL-1R8, subsequently activating multiple intracellular switches to block inflammation, which includes inhibition of NLRP3 inflammasome and activation of cytokine signal transduction inhibitor 3 (SOCS3). Thus, rhIL-37 limits the inflammatory response caused by MSU crystals. Meanwhile, a comparative study [55] found that the expression of IL-1, IL-6, and TNF-α in the acute gouty arthritis (AGA) group were significantly higher than those in the non-acute gouty arthritis (NAGA) group and the healthy control (HC) group; however, the anti-inflammatory IL-37 and TGF-β1 as well as IL-10 in the NAGA group were significantly higher than those in the AGA group and the HC group. This study showed that IL-37 plays a special anti-inflammatory factor role in AGA by inhibiting the production of pro-inflammatory cytokines. In some AGA, elevated expression of IL-37 is likely associated with some negative feedback mechanisms that inhibit excessive inflammation, and this conclusion is also consistent with the self-limiting nature of MSU crystal-induced acute episodes of gout.
The immune cells
Neutrophils
Neutrophil invasion and activation is an important process of gout acute inflammation. Neutrophils engulf MSU crystals and induce the production of phagolysosomes, causing the production of reactive oxygen species, the release of inflammatory factors (IL-1β, IL-6, TNF-α), and the appearance of arachidonic acid products. Membrane damage can cause neutrophil death, lysosomal contents, and newly synthesized inflammatory mediators diffuse into local tissues, triggering an acute attack of arthritis. MSU crystals can also activate locally associated immunoglobulins, causing rapid aggregation of neutrophils, triggering an acute attack of gout. As inflammation reaches a peak, neutrophils are stimulated to form neutrophil extracellular trapping nets [56], then capture and degrade inflammatory factors and rapidly attenuate the inflammatory response, which may explain the clinical manifestations of acute and rapid remission of acute gouty arthritis.
Monocytes/macrophages (THP-1)
THP-1 inflammatory response induced by MSU crystal stimulation has also been confirmed in animal gout models [57]. MSU crystals activate THP-1 through the Toll-like receptor pathway and NALP3 inflammasomes, thereby activating caspase-1, converting precursors IL-1β and IL-18 into active forms, and this can also increase expression of the COX-2 and NF-κΒ (p65) on the THP-1, stimulating cells to secrete TNF-α and causing inflammatory response [32, 58]. But some differentiated macrophages can alleviate inflammation induced by MSU crystals by secreting transforming growth factor-β1 (TGF-β1). In short, MSU crystals interact with macrophages to trigger inflammation and induce neutrophil and monocyte infiltration to expand the inflammatory response. However, with the involvement of various mechanisms such as TGF-β1, macrophages can induce spontaneous remission of inflammation. It is also consistent with the feature that clinical gout can relieve itself.
T lymphocytes
CD4+T cell subsets in peripheral blood can differentiate into helper T cells (Th)1, Th2, Th17, and regulatory T (Treg), of which Th1 and Th2 cells are considered to be closely related to the onset of gout. Th1 cells mediate cellular immunity mainly by secreting cytokines such as IFN-γ, IL-2, and IL-18. Th2 cells mainly secrete cytokines such as IL-4 and IL-10 to exert important anti-inflammatory effects. A study found that Th1 and the ratio of Th1/ Th2 in peripheral blood of patients with acute gout were significantly increased, but Th2 cells were significantly decreased, indicating the imbalance of Th1/Th2 cell subsets may run through the onset of gouty arthritis [59]. Before that, another study also found that Th17 and Treg cells are closely related to the pathogenesis of gout. Th17 cells of patients with acute gout were increased accompanied by a decrease in Treg cells, indicating that the imbalance of Th17/Treg plays an important role in the acute attack of gout [60]. In the presence of IL-1β and IL-18, MSU can differentiate and activate Th17, thereby producing IL-17 to promote the inflammatory response [44]. Activated receptors of NF-κΒ ligands (RANKL) on T cells can differentiate osteoclasts and lead to bone destruction and gouty arthritis [61]. In contrast, Treg cells can inhibit osteoclastogenesis by secreting IL-4, IL-10, and TGF-β1, reducing gout bone damage [62]. Therefore, as previously mentioned, the release of IL-17 by Th17 may be a potential pathological mechanism for acute gout attacks (Fig. 4).

The immune cells in gout MSU crystals can stimulate neutrophils and monocytes/macrophages to produce IL-1β, IL-6, and TNF-α, stimulate Th17 cells to produce IL-17. Th1 cells mediate cellular immunity mainly by secreting cytokines such as IFN-γ, IL-2, and IL-18. They synergistically promote the occurrence of gout inflammation. In addition, NF-κΒ activated receptor ligands (RANKL) on T cells can differentiate osteoclasts, leading to bone destruction and gouty arthritis. However, Treg cells can produce IL-4, IL-10, and TGF-β1 to reduce gout bone damage, and Th2 cells mainly secrete cytokines such as IL-4 and IL-10 to exert important anti-inflammatory effects
Relief of acute gout
The relief mechanism of acute gout is a complex process involving multiple factors. It is still the focus and difficulty of current research. The mechanism is currently believed to have the following points. First, the upregulation of anti-inflammatory factors: in the early and middle stages of inflammation, the levels of inflammatory cells and IL-1β, IL-6, and TNF-α in synovial fluid are significantly increased, while in the late stage of inflammation, anti-inflammatory factors TGF-β1 levels increased significantly [63]. TGF-β1 is considered to be an important anti-inflammatory factor in spontaneous remission of gout. Second, with the progress of acute gout inflammation, infiltrating monocyte-macrophages differentiate into inflammatory M1-like macrophages, rather than M2-like macrophages, and the increased production of TGF-β1 may be related to the enhanced devoured ability of monocytes/macrophages to apoptotic neutrophils. In addition, elevated IL-10 blocks the TNF-α production of macrophages [57]. In short, increased production of anti-inflammatory factors plays an important role in the spontaneous remission of gout. Third, the sterilized environment in the joint cavity during acute gout prevents the TLR/NF-κΒ pathway from being continuously activated, limiting the production of the pro-IL-1β [12]. Fourth, neutrophils aggregate to form neutrophil extracellular trapping nets, phagocytize apoptotic cells, release TGF-β1, capture and degrade inflammatory factors, so limiting inflammatory cell infiltration and pro-inflammatory effects [56]. With the in-depth study of the self-limiting relief mechanism of acute gout, more new targets for the treatment of acute gout can be discovered.
Summary and prospect
The acute onset of gout inflammation is the MSU crystal-mediated inflammatory cascade amplification, by the effects of various cytokines and inflammatory bodies throughout the process. As a pathogen-associated molecule, MSU crystal involved in the induction of acute inflammation of gout through TLRs, stimulating the production of NLRP3 inflammasome, interleukin-like factors, and TNF-α, regulating the development of inflammation. With the in-depth study about the mechanism of gout inflammation, the corresponding treatment measures for gout inflammation have also increased, especially for the targeted biologic treatment of refractory gout cases, such as tumor necrosis factor alpha antagonists, IL-1R antagonist (anakinra), human monoclonal antibody of human IL-1β (kananazumab), and human monoclonal antibody of human IL-6R (tocilizumab) have been validated in clinical trials for a variety of diseases [64, 65]. Studies have also found that the interaction of NLPR3 heterodimers with NLPR3 can negatively regulate the activation of NLPR3 and thus control inflammation [66]. Therefore, further research is necessary for the inflammatory mechanisms and for the future inflammatory targets’ management of gout.
Abbreviations
- ASC:
-
apoptosis-associated speck-like protein
- AGA:
-
acute gouty arthritis
- ATP:
-
adenosine triphosphate;
- CAM-1:
-
cellular adhesion molecule 1
- CIA:
-
collagen arthritis
- CLIC:
-
intracellular chloride channel protein
- DAMP:
-
dangerous-associated molecular patterns
- GM-CSF:
-
granulocyte-macrophage stimulating factor
- HC:
-
healthy control
- IL:
-
interleukin
- LRR:
-
leucine-rich repeat domain
- LPS :
-
bacterial lipopolysaccharide
- MAVS:
-
mitochondrial antiviral signaling protein
- MSU:
-
monosodium urate monohydrate
- MIP-2:
-
macrophage inflammatory protein 2
- Mfn2:
-
Mitochondrial fusion protein 2
- NF-κB:
-
activate nuclear transcription factor
- NLR:
-
NOD-like receptors
- NAGA:
-
non-acute gouty arthritis
- PRR:
-
pattern recognition receptor
- PAMP:
-
pathogen-associated molecular patterns
- PYD:
-
pyrin domain
- P2X7:
-
P2X purine receptor 7
- RANKL :
-
NF-κΒ ligands
- ROS:
-
active oxygen
- SOCS3:
-
signal transduction inhibitor 3
- TGF-β1 :
-
transforming growth factor-β1
- TNF-α:
-
tumor necrosis factor alpha
- TLR:
-
Toll-like receptor
References
Dalbeth N, Phipps-Green A, Frampton C, Neogi T, Taylor WJ, Merriman TR (2018) Relationship between serum urate concentration and clinically evident incident gout: an individual participant data analysis. Ann Rheum Dis 77(7):1048–1052
Dalbeth N, House ME, Aati O et al (2015) Urate crystal deposition in asymptomatic hyperuricaemia and symptomatic gout: a dual energy CT study. Ann Rheum Dis 74:908–911
So AK, Martinon F (2017) Inflammation in gout: mechanisms and therapeutic targets. Nat Rev Rheumatol 13(11):639–647
Scott P, Ma H, Viriyakosol S, Terkeltaub R, Liu-Bryan R (2006) Engagement of CD14 mediates the inflammatory potential of monosodium urate crystals. J Immunol 177(9):6370–6378
Barbero F, Russo L, Vitali M, Piella J, Salvo I, Borrajo ML, Busquets-Fité M, Grandori R, Bastús NG, Casals E, Puntes V (2017) Formation of the protein corona: the Interface between nanoparticles and the immune system. Semin Immunol 34:52–60
Pascual E (1994) Hyperuricemia and gout. Curr Opin Rheumatol 6(4):454–458
Renaudin F, Sarda S, Campillo-Gimenez L, Séverac C, Léger T, Charvillat C, Rey C, Lioté F, Camadro JM, Ea HK, Combes C (2019) Adsorption of proteins on m-CPPD and urate crystals inhibits crystal-induced cell responses: study on albumin-crystal interaction. J Funct Biomater 10(2):18
Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G, Rubartelli A (2008) ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1β and IL-18 secretion in an autocrine way. Proc Natl Acad Sci U S A 105:8067–8072
Scanu A, Luisetto R, Oliviero F, Gruaz L, Sfriso P, Burger D, Punzi L (2015) High-density lipoproteins inhibit urate crystal-induced inflammation in mice. Ann Rheum Dis 74:587–594
Rock KL, Kataoka H, Lai JJ (2013) Uric acid as a danger signal in gout and its comorbidities. Nat Rev Rheumatol 9(1):13–23
Joosten LA, Abdollahi-Roodsaz S, Dinarello CA et al (2016) Toll-like receptors and chronic inflammation in rheumatic diseases: new developments. Nat Rev Rheumatol 12(6):344–357
Narayanan KB, Park HH (2015) Toll/interleukin-1 receptor (TIR) domain-mediated cellular signaling pathways. Apoptosis. 20(2):196–209
Liu-Bryan R, Scott P, Sydlaske A, Rose DM, Terkeltaub R (2005) Innate immunity conferred by toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum 52(9):2936–2946
Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, Akira S, Rock KL (2006) MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest 116(8):2262–2271
Qi W, Cheng X-S (2015) Research advances in NLRP3 inflammasome. Basic Clin Med 15(1):117–121
Elliott EI, Sutterwala FS (2015) Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev 265(1):35–52
Samways DS, Li Z, Egan TM (2014) Principles and properties of ion flow in P2X receptors. Front Cell Neurosci 8:6
Di A, Xiong S, Ye Z et al (2018) The TWIK2 potassium efflux channel in macrophages mediates NLRP3 inflammasome-induced inflammation. Immunity 49(1):56–65.e4
Triantafilou K, Hughes TR, Triantafilou M, Morgan BP (2013) The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J Cell Sci 126(Pt 13):2903–2913
Laudisi F, Spreafico R, Evrard M, Hughes TR, Mandriani B, Kandasamy M, Morgan BP, Sivasankar B, Mortellaro A (2013) Cutting edge: the NLRP3 inflammasome links complement-mediated inflammation and IL-1β release. J Immunol 191(3):1006–1010
Asgari E, Le Friec G, Yamamoto H et al (2013) C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood. 122(20):3473–3481
Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G (2013) K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 38(6):1142–1153
Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, Robertson AAB, Cooper MA, Graf T, Hornung V (2016) Human monocytes engage an alternative inflammasome pathway. Immunity. 44(4):833–846
Groß CJ, Mishra R, Schneider KS, Médard G, Wettmarshausen J, Dittlein DC, Shi H, Gorka O, Koenig PA, Fromm S, Magnani G, Ćiković T, Hartjes L, Smollich J, Robertson AAB, Cooper MA, Schmidt-Supprian M, Schuster M, Schroder K, Broz P, Traidl-Hoffmann C, Beutler B, Kuster B, Ruland J, Schneider S, Perocchi F, Groß O (2016) K+ efflux-independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity. 45(4):761–773
Courbet A, Bec N, Constant C, Larroque C, Pugniere M, el Messaoudi S, Zghaib Z, Khier S, Deleuze-Masquefa C, Gattacceca F (2017) Imidazoquinoxaline anticancer derivatives and imiquimod interact with tubulin: characterization of molecular microtubule inhibiting mechanisms in correlation with cytotoxicity. PLoS One 12(8):e0182022
Zghaib Z, Guichou JF, Vappiani J, Bec N, Hadj-Kaddour K, Vincent LA, Paniagua-Gayraud S, Larroque C, Moarbess G, Cuq P, Kassab I, Deleuze-Masquéfa C, Diab-Assaf M, Bonnet PA (2016) New imidazoquinoxaline derivatives: synthesis, biological evaluation on melanoma, effect on tubulin polymerization and structure-activity relationships. Bioorg Med Chem 24(11):2433–2440
Haneklaus M, O’Neill LA, Coll RC (2013) Modulatory mechanismscontrolling the NLRP3 inflammasome in inflammation: recent developments. Curr Opin Immunol 25(1):40–45
Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J (2010) Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 11(2):136–140
Chen J, Chen ZJ (2018) PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature. 564(7734):71–76
Martin WJ, Walton M, Harper J (2009) Resident macrophages initiating and driving inflammation in a monosodium Urate monohydrate crystal-induced murine peritoneal model of acute gout. Arthritis Rheum 60(1):281–289
Hoffman HM, Scott P, Mueller JL, Misaghi A, Stevens S, Yancopoulos GD, Murphy A, Valenzuela DM, Liu-Bryan R (2010) Role of the leucine-richrepeat domain of cryopyrin/NLRP3 in monosodium urate crystai-induced inflammation in mice. Arthritis Rheum 62(7):2170–2179
Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J (2006) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 440(7081):237–241
Pisetsky DS, Ward MM (2012) Advances in the treatment of inflammatory arthritis. Best Pract Res Clin Rheumatol 26(2):251–261
Bauernfeind F, Niepmann S, Knolle PA, Hornung V (2016) Aging-associated TNF production primes inflammasome activation and NLRP3-related metabolic disturbances. J Immunol 197(7):2900–2908
Yokose K, Sato S, Asano T, Yashiro M, Kobayashi H, Watanabe H, Suzuki E, Sato C, Kozuru H, Yatsuhashi H, Migita K (2018) TNF-α potentiates uric acid-induced interleukin-1β (IL-1β) secretion in human neutrophils. Mod Rheumatol 28(3):513–517
Amaral FA, Bastos LF, Oliveira TH et al (2016) Transmembrane TNF-α is sufficient for articular inflammation and hypernociception in a mouse model of gout. Eur J Immunol 46(1):204–211
Cabrera SM, Wang X, Chen YG, Jia S, Kaldunski ML, Greenbaum CJ, the Type 1 Diabetes TrialNet Canakinumab Study Group, Mandrup-Poulsen T, the AIDA Study Group, Hessner MJ (2016) Interleukin-1 antagonism moderates the inflammatory state associated with type 1 diabetes during clinical trials conducted at disease onset. Eur J Immunol 46(4):1030–1046
Busso N, So A (2010) Mechanisms of inflammation in gout. Arthritis Res Ther 12(2):206
Sutton C, Brereton C, Keogh B, Mills KHG, Lavelle EC (2006) A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med 203(7):1685–1691
Janssen CA, Oude Voshaar MAH, Vonkeman HE, Jansen TLTA, Janssen M, Kok MR, Radovits B, van Durme C, Baan H, van de Laar MAFJ (2019) Anakinra for the treatment of acute gout flares: a randomized, double-blind, placebo-controlled, active-comparator, non-inferiority trial. Rheumatology (Oxford) 58:1344–1352
Kim KW, Kim BM, Lee KA et al (2019) Reciprocal interaction between macrophage migration inhibitory factor and interleukin-8 in gout. Clin Exp Rheumatol 37(2):270–278
Liu R, Aupperle K, Terkeltaub R (2001) Src family protein tyrosine kinase signaling mediates monosodium urate crystal-induced IL-8 expression by monocytic THP-1 cells. J Leukoc Biol 70(6):961–968
Liu R, O’Connell M, Johnson K et al (2000) Extracellular signal-regulated kinase 1/extracellular signal-regulated kinase 2 mitogen-activated protein kinase signaling and activation of activator protein 1 and nuclear factor kappaB transcription factors play central roles in interleukin-8 expression stimulated by monosodium urate monohydrate and calcium pyrophosphate crystals in monocytic cells. Arthritis Rheum 43(5):1145–1155
Conforti-Andreoni C, Spreafico R, Qian HL, Riteau N, Ryffel B, Ricciardi-Castagnoli P, Mortellaro A (2011) Uric acid-driven Th17 differentiation requires inflammasome-derived IL-1 and IL-18. J Immunol 187(11):5842–5850
Mills KH, Dungan LS, Jones SA et al (2013) The role of inflammasome-derived IL-1 in driving IL-17 responses. J Leukoc Biol 93(4):489–497
Liu Y, Zhao Q, Yin Y, McNutt MA, Zhang T, Cao Y (2018) Serum levels of IL-17 are elevated in patients with acute gouty arthritis. Biochem Biophys Res Commun 497(3):897–902
Johnson JL, Jones MB, Cobb BA (2018) Polysaccharide-experienced effector T cells induce IL-10 in FoxP3+ regulatory T cells to prevent pulmonary inflammation. Glycobiology. 28(1):50–58
Chen S, Chen B, Wen Z, Huang Z, Ye L (2017) IL-33/ST2-mediated inflammation in macrophages is directly abrogated by IL-10 during rheumatoid arthritis. Oncotarget. 8(20):32407–32418
Chen YH, Hsieh SC, Chen WY, Li KJ, Wu CH, Wu PC, Tsai CY, Yu CL (2011) Spontaneous resolution of acute gouty arthritis is associated with rapid induction of the anti-inflammatory factors TGF-β1, IL-10 and soluble TNF receptors and the intracellular cytoldne negative regulators CIS and SOCS3. Ann Rheum Dis 70(9):1655–1663
Schreiber S, Heinig T, Thiele HG, Raedler A (1995) Immuno-regulatory role of interleukin-10 in patients with inflammatory bowel disease. Gastroenterology. 108(5):1434–1444
Zhuang X, Wu B, Li J, Shi H, Jin B, Luo X (2017) The emerging role of interleukin-37 in cardiovascular diseases. Immun Inflamm Dis 5(3):373–379
Eisenmesser EZ, Gottschlich A, Redzic JS, Paukovich N, Nix JC, Azam T, Zhang L, Zhao R, Kieft JS, The E, Meng X, Dinarello CA (2019) Interleukin-37 monomer is the active form for reducing innate immunity. Proc Natl Acad Sci U S A 116(12):5514–5522
Nold MF, Nold-Petty CA, Zepp JA et al (2010) IL-37 is a fundamental inhibitor of innate immunity. Nat Immunol 11(11):1014–1022
Liu L, Xue Y, Zhu Y, Xuan D, Yang X, Liang M, Wang J, Zhu X, Zhang J, Zou H (2016) Interleukin 37 limits monosodium urate crystal-induced innate immune responses in human and murine models of gout. Arthritis Res Ther 18(1):268
Zeng M, Dang W, Chen B, Qing Y, Xie W, Zhao M, Zhou J (2016) IL-37 inhibits the production of proinflammatory cytokines in MSU crystal-induced inflammatory response. Clin Rheumatol 35:2251–2258
Alvarez-Soria MA, Herrero-Beaumont G, Sánchez-Pernaute O et al (2008) Diacerein has a weak effect on the catabolic pathway of human osteoarthritis synovial fibroblast—comparison to its effects on osteoarthritic chondrocytes. Rheumatology(Oxford). 47(5):627–633
Martin WJ, Shaw O, Liu X, Steiger S, Harper JL (2011) Monosodium urate monohydrate crystal-recruited noninflammatory monocytes differentiate into M1-like proinflammatory macrophages in a peritoneal murine model of gout. Arthritis Rheum 63:1322–1332
Luo C (2015) Distinct impact of uric acid crystals and high uric acid on human monocytes/macrophages THP-1. Shantou University R589.7: 1–77
Yang H, Yang X, Yufeng Q et al (2018) Study on the expression and significance of Th1/Th2 cells in the blood of patients with primary gouty arthritis. Chin J Rheumatol 22(11):731–736
Yang H, Yang X, Xiaowu Z et al (2016) Role of Thl7/Treg cell balance in the pathogenesis of primary gout arthritis. Chin J Rheumatol 20(8):520–525
Lee SJ, Nam KI, Jin HM, Cho YN, Lee SE, Kim TJ, Lee SS, Kee SJ, Lee KB, Kim N, Park YW (2011) Bone destruction by receptor activator of nuclear factor kappaΒ ligand-expressing T cells in chronic gouty arthritis. Arthritis Res Ther 13(5):R164
Luo CY, Wang L, Sun C, Li DJ (2011) Estrogen enhances the functions of CD4(+)CD25(+) Foxp3(+) regulatory T cells that suppress osteoclast differentiation and bone resorption in vitro. Cell Mol Immunol 8(1):50–58
Scanu A, Oliviero F, Ramonda R, Frallonardo P, Dayer JM, Punzi L (2012) Cytokine levels in human synovial fluid during the different stages of acute gout: role of transforming growth factor β1 in the resolution phase. Ann Rheum Dis 71(4):621–624
Satoh T, Otsuka A, Contassot E, French LE (2015) The inflammasome and IL-1β: implications for the treatment of inflammatory diseases. Immunotherapy. 7(3):243–254
Mokuda S, Kanno M, Takasugi K et al (2014) Tocilizumab improved clinical symptoms of a patient with systemic tophaceous gout who had symmetric polyarthritis and fever: an alternative treatment by blockade of interleukin-6 signaling. SAGE Open Med Case Rep 2:2050313X13519774
Yang CS, Kim JJ, Kim TS, Lee PY, Kim SY, Lee HM, Shin DM, Nguyen LT, Lee MS, Jin HS, Kim KK, Lee CH, Kim MH, Park SG, Kim JM, Choi HS, Jo EK (2015) Small heterodimer partner interacts with NLRP3 and negatively regulates activation of the NLRP3 inflammasome. Nat Commun 6:6115
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81270577), the Science and Technology Innovation Foundation of Shenzhen, China (No. JCYJ20160427191026117), the Science and Technology Innovation Foundation of Baoan, Shenzhen, China (No. 2018JD237, No. 2016CX191), and the Construction Units of Key Specialties in Clinical Medicine, Baoan District, Shenzhen, China (No. 8, 214-2018, Health Commission of Baoan, Shenzhen City).
Author information
Authors and Affiliations
Contributions
Literature review: Meimei Wu and Chengshan Guo; Drafting of manuscript: Meimei Wu, Ye Tian, and Qianqian Wang. Revision of manuscript: Chengshan Guo and Meimei Wu. All authors have read and approved the final submitted manuscript.
Corresponding author
Ethics declarations
Disclosures
None.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wu, M., Tian, Y., Wang, Q. et al. Gout: a disease involved with complicated immunoinflammatory responses: a narrative review. Clin Rheumatol 39, 2849–2859 (2020). https://doi.org/10.1007/s10067-020-05090-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-020-05090-8