
Abstract
Objective
Systemic lupus erythematosus (SLE) is a complex autoimmune disease characterized by autoantibody production and multi-system involvement, but the etiology is largely unclear. This study aimed to elucidate candidate genes and pathways involved in SLE.
Methods
Three original datasets GSE72509, GSE20864, and GSE39088 were downloaded from Gene Expression Omnibus (GEO) and the data were further integrated and analyzed. Subsequently, differentially expressed genes (DEGs) between SLE patients and healthy people were identified. And then we performed gene ontology (GO) function and pathway enrichment analyses of common DEGs, and constructed a protein-protein interaction (PPI) network with STRING database. Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) was carried out to validate the expression levels of candidate genes in blood samples from SLE patients and healthy controls.
Results
In total, 321 common DEGs were identified in SLE patients compared with healthy controls, including 231 upregulated and 90 downregulated genes. GO function analysis revealed that 321 common DEGs were mainly enriched in innate immune response, defense response, cytokine-mediated signaling pathway, response to interferon-alpha, and I-kappaB kinase/NF-kappaB signaling. Additionally, pathway enrichment analysis indicated that DEGs were mainly enriched in several signaling pathways associated with immune system and apoptosis, including RIG-I-like receptor signaling pathway, antigen processing and presentation, and p53 signaling pathway. The expression levels of candidate genes RPL26L1, FBXW11, FOXO1, and SMAD7 were validated by RT-qPCR analysis.
Conclusions
The four hub genes including RPL26L1, FBXW11, FOXO1, and SMAD7 may play key roles in the pathogenesis and development of SLE. RIG-I-like receptor signaling pathway, antigen processing and presentation pathway, and p53 signaling pathway may be closely implicated in SLE pathogenesis. Collectively, these results may provide valuable novel markers or targets for the diagnosis and treatment of SLE.
Key Points • Integrated bioinformatics analysis of three profile datasets based on SLE patients and healthy controls was performed and 321 common DEGs were identified. • The 321 common DEGs were mainly enriched in biological processes related to immune responses and inflammatory responses, including innate immune response, defense response, cytokine-mediated signaling pathway, response to interferon-alpha, I-kappaB kinase/NF-kappaB signaling, whereas the three most significant cellular components were oxidoreductase complex, AIM2 inflammasome complex, and ubiquitin ligase complex. • KEGG pathway enrichment analysis indicated that common DEGs were mainly enriched in several signaling pathways associated with immune system and apoptosis, including RIG-I-like receptor signaling pathway, antigen processing and presentation, and p53 signaling pathway. • Candidate genes RPL26L1, FBXW11, FOXO1, and SMAD7 may be closely involved in the pathogenesis and development of SLE and may provide valuable novel markers or targets for the diagnosis and treatment of SLE. |
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although systemic lupus erythematosus (SLE) is generally recognized as a complex autoimmune disease induced by environmental and genetic factors [1], the detailed etiology is still obscure. SLE can affect almost any organs and tissues, resulting in highly diversified clinical manifestations, mostly including fever, rashes, oral ulcer, arthritis, vasculitis, and life-threatening nephritis [1]. It is more prevalent in African, Asian, Hispanic, and American, and the prevalence of SLE in China is about 30 per 100,000 [2]. Due to the high morbidity and mortality, it is critically important and highly demanded to uncover the underlying molecular mechanisms and discover the reliable biomarkers for early diagnosis and effective therapy.
Over the past five decades, a lot of efforts have been made in the understanding of the pathogenesis and development of SLE. From a cellular perspective, lymphocytes were considered to play a central role in the pathogenesis of SLE. Aberrantly activated T cells or imbalance of T cell subsets (e.g., decreased production of Treg cells and increased production of Th 17 cells) [3, 4] mediate inflammatory responses and activate B cells to differentiate and produce more autoantibodies, contributing to pathogenesis and development of SLE. Recently, the role of innate immune cells in the pathogenesis of SLE has attracted more attention. Plasmacytoid dendritic cells (pDCs) have been found to infiltrate in the renal and skin lesions of SLE patients [5]. Plasmacytoid dendritic cells produce a large number of type I IFN that is often found elevated in SLE patients [6]. Type I IFNs, as proinflammatory cytokines, can enhance immune responses by modulating T cell and B cell function [7,8,9]. Thus, pDCs have been suggested to be implicated to play a key role in the pathogenesis of SLE. On the other hand, neutrophils have been reported with decreased clearance of apoptotic material and increased synthesis and release of various inflammatory cytokines in SLE [10]. Neutrophils undergo a new cell death process to release neutrophil extracellular traps (NETs), a network of chromatin filaments coated with histones, proteases, and cytosolic proteins [11]. The release of NETs provides a source of autoantigens and enhances inflammatory and immune responses. These observations have provided massive evidence on the contribution of neutrophil to the pathogenesis of SLE. However, the underlying molecular mechanisms on the pathogenesis of SLE remain to be deeply elucidated.
With the wide application of gene detection technique, more and more microarray data profiling studies on SLE were performed and hundreds of differentially expressed genes (DEGs) were identified. Integrating and re-analyzing these data by bioinformatics methods can provide new and valuable ideas for understanding the molecular mechanisms and identifying reliable diagnostic and therapeutic targets of SLE. Therefore, in this study, we downloaded three original datasets GSE72509, GSE20864, and GSE39088 from NCBI-GEO, including 198 SLE patients and 108 healthy controls from three different racial populations. We integrated and re-analyzed the data and identified 321 common DEGs between SLE patients and healthy people. We performed gene ontology (GO) function and pathway enrichment analyses of the common DEGs with DAVID, Gene Ontology, and KEGG PATHWAY, and constructed a protein-protein interaction (PPI) network of DEGs. Finally, we validated the expression levels of these candidate genes by reverse transcription-quantitative polymerase chain reaction (RT-qPCR). The results of this study may help to improve our comprehension of the underlying molecular mechanisms of SLE and provide potential targets for diagnosis and therapy of SLE.
Materials and methods
GEO data information
Whole blood gene expression profile of GSE72509, GSE20864, and GSE39088 from SLE patients and healthy controls was obtained from NCBI-GEO (available online: https://www.ncbi.nlm.nih.gov/geo). The GSE72509 was a RNA-seq data sequenced by Illumina HiSeq 2500. It contained 99 SLE patients and 18 healthy controls [12]. The GSE20864 was a microarray data generated by Hitachisoft AceGene Human Oligo Chip 30K 1 Chip Version. It included 21 SLE patients and 45 healthy controls [13]. And the GSE39088 was generated by Affymetrix Human Genome U133 Plus 2.0 Array. The 78 non-treatment SLE patients and 46 healthy controls were chosen to be analyzed [14, 15]. The datasets represented three different areas: the GSE72509 conducted in the USA, the GSE20864 generated in Japan, and the GSE39088 data from Belgium.
Identification of differentially expressed genes
The expression data of GSE72509, GSE20864, and GSE39088 were directly imported into Qlucore Omics Explorer 3.2 (QOE) software. To identify the DEGs between SLE patients and healthy controls, p < 0.01 (two-group comparisons, two-tailed) was considered significant between the gene expression differences. The heat maps were drawn using the QOE with the normalization of mean = 0 and variance = 1 for each genes.
Gene ontology and pathway enrichment analyses
GO term enrichment analysis and network analysis were using Cytoscape ClueGO plugin with default setting. Kyoto Encyclopedia of Genes and Genomes (KEGG) is a collection of online databases for the systematic analysis of gene functions in light of networks of genes and molecules. To identify enrichment pathways of DEG, the data were analyzed using Database for Annotation, Visualization and Integrated Discovery (DAVID) [16, 17]. The thresholds were Count (minimum count) ≥ 2, ESAE (modified Fisher exact p value) ≥ 0.1 (default).
Analysis of protein-protein interaction network
To further investigate the underlying molecular mechanisms of SLE, a protein-protein interaction (PPI) network for the DEGs was constructed by using the STRING database (http://www.string-db.org/) (the confidence score cutoff was 900) and then visualized by Network Analyst (http://www.networkanalyst.ca) [16]. Finally, the nodes with higher degrees of interaction were considered as candidate genes.
RT-qPCR to confirm candidate genes
To confirm the expression of candidate genes in SLE patients and healthy controls, whole blood RT-qPCR analysis was performed. A total of 5 ml fresh whole blood samples were obtained from patients with SLE (n = 14; all female; age range, 18–54 years, SLEDAI (mean ± SD), 12.93 ± 4.87, new onset: previously treated patients = 2:12, patients with lupus nephritis, n = 11) and normal healthy controls (n = 12; 3 male and 9 female; age range, 24–30 years). All the fourteen patients were recruited from the Third Affiliated Hospital of Southern Medical University (Guangzhou, China) and informed written consent requested. The protocol was approved by the Ethics Committee of The Third Affiliated Hospital of Southern Medical University (No. 201603003). Total white cell RNA was extracted using TRIzol reagent (Vazyme, Nanjing, China). The first strand complementary DNAs (cDNAs) were synthesized using the PrimeScript RT reagents Kit (Perfect Real Time, Takara) according to the manufacturer’s instructions. And then, RT-qPCR analysis was performed in triplicate in an ABI 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control for quantification of target gene expression. The following PCR primers were used: FBXW11, forward, 5′-CCGACTCGGTGATTGAGGAC-3′, and reverse, 5′-CCGATGTTCCCCACATCCAA-3′; FOXO1, forward, 5′-AACCTGTCCTACGCCGACCTCA-3′, and reverse, 5′-GCTCGGCTTCGGCTCTTAGCAA-3′; SMAD7, forward, 5′-GTGGCATACTGGGAGGAGAA-3′, and reverse, 5′-GATGGAGAAACCAGGGAACA-3′; GAPDH, forward, 5′-TGGACTCCACGACGTACTCAG-3′, and reverse, 5′-CGGGAAGCTTGTCATCAATGGAA-3′; PLCB1, forward, 5′-GTGTCCGACAGCCTCAAGAA-3′, and reverse, 5′-CCGATGTTCCCCACATCCAA-3′; SIN3A, forward, 5′-ACCATGCAGTCAGCTACGG-3′, and reverse, 5′-CACCGCTGTTGGGTGATGA-3′, RPL26L1, forward, 5′-TTCAATCCCTTCGTTACCTCGG-3′, and reverse, 5′-TAGTGTCCTCGAACTACCTGG-3′. To quantify gene expression, 2-step qRT-PCR was performed using hot start Taq at 95 °C (15 s), with annealing and extension at 60 °C (60 s) for 40 cycles, followed by a melting curve analysis. All qRT-PCR data were analyzed using the 2−ΔΔCt method [17]. All the statistics were analyzed with SPSS version 20.0. RT-qPCR data were expressed as the mean ± standard error of mean (SEM). Statistical significance was determined using an independent sample t test and p < 0.05 was considered to be statistically significant.
Results
Identification of DEGs in SLE
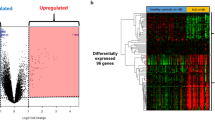
Three gene expression data containing SLE patients and healthy controls were analyzed with two-group comparisons (p < 0.01) and then 4461, 3400, and 4212 DEGs from GSE72509, GSE20864, and GSE39088 were identified respectively. The heat maps of these DEGs were shown in Fig. 1a. After integration of these DEGs by employing bioinformatics analysis, a total of 321 common DEGs were identified in SLE patients compared with healthy controls (Fig. 1b), including 231 upregulated genes and 90 downregulated genes (Supplementary Table 1).

a The heat maps of the three profile datasets GSE39088, GSE72509, and GSE20864. The profile dataset GSE39088 that generated from Belgium contained 78 SLE patients and 46 healthy controls. The profile dataset GSE72509 which was conducted in the USA contained 99 SLE patients and 18 healthy controls. The profile dataset GSE20864 included 21 SLE patients and 45 healthy controls from Japan. b Identification of 321 common DEGs from the three datasets GSE72509, GSE20864, and GSE39088. Different color areas represented different datasets. The cross areas meant the common DEGs. DEGs were identified with classical t test and statistically significant DEGs were defined with p < 0.01 as the cutoff criterion (DEGs, differentially expressed genes)
Gene ontology analysis of DEGs in SLE
The DEGs were categorized into three function groups: cellular component group, molecular function group, and biological process group. GO analysis showed that the 321 common DEGs were significantly enriched in some biological processes related to SLE, for example, innate immune response, defense response, cytokine-mediated signaling pathway, response to interferon-alpha, and I-kappaB kinase/NF-kappaB signaling. In the cellular component group, three most significantly enriched GO terms of the 321 common DEGs were oxidoreductase complex, AIM2 inflammasome complex, and ubiquitin ligase complex. In the molecular function group, the most significantly enriched GO term of the 321 common DEGs was single-stranded RNA binding (Fig. 2 and Supplementary Table 2).

Significantly enriched GO terms of DEGs in SLE based on their GO functions. The 321 common DEGs were categorized into three function groups: cellular component group, molecular function group, and biological process group. Each node represents a GO term. The edges reflect the relationships between the terms based on the similarity of their associated genes. The larger node size and deeper color indicate greater significance of the enrichment (GO, gene ontology; DEGs, differentially expressed genes; SLE, systemic lupus erythematosus)
Signaling pathway enrichment analysis of DEGs in SLE
Signaling pathway enrichment analysis of DEGs was performed with a knowledge base-KEGG PATHWAY. The detailed results of pathway enrichment analysis were shown in Fig. 3a and Supplementary Table 3. The 321 common DEGs were mainly enriched in some signaling pathways associated with immune system and apoptosis including RIG-I-like receptor signaling pathway, antigen processing and presentation, and p53 signaling pathway. The details of these three important signaling pathways were presented in Fig. 3b, c, and d. The DEGs were also enriched in infectious diseases including influenza A and herpes simplex infection.

Significantly enriched KEGG pathway terms of DEGs in SLE. The details of the three important signaling pathways—RIG-I-like receptor signaling pathway, p53 signaling pathway, and antigen processing and presentation—are presented (DEGs, differentially expressed genes; SLE, systemic lupus erythematosus)
A protein-protein interaction network for DEGs
The PPI network for the 321 common DEGs was constructed by employing the STRING online database and Network Analyst. It contained 1713 nodes and 2512 edges (Fig. 4). Among the nodes, 24 node genes were identified as candidate genes with an interaction degree > 28 (each node had more than 28 connections/interactions) (Supplementary Table 4). Among the candidate genes, RPL26L1 showed the highest node degrees, followed by SIN3A, PLCB1, FOXO1, SMAD7, and FBXW11.

A protein-protein interaction (PPI) network for DEGs
Validation of candidate genes by RT-qPCR
To validate these major results, we selectively performed RT-qPCR analysis of six random important DEGs including RPL26L1, FOXO1, SMAD7, FBXW11, PLCB1, and SIN3A in 14 SLE patients and 12 healthy controls. The relative mRNA expression levels of FOXO1 and SMAD7 were significantly lower in blood samples from SLE patients when compared with those from healthy controls. The expression levels of RPL26L1 and FBXW11 were significantly higher in SLE group than healthy control group. However, there were no significant differences in the relative mRNA expression levels of PLCB1 and SIN3A between SLE patients and healthy people (Fig. 5).

Relative mRNA expression levels of RPL26L1, FBXW11, FOXO1, SMAD7, SIN3A, and PLCB1 in blood samples from SLE patients and healthy controls, as determined using RT-qPCR. (n = 12 for healthy controls, n = 14 for SLE patients. Mean ± SEM are plotted; *p < 0.05; **p < 0.01; ***p < 0.001)
Discussion
Systemic lupus erythematosus is a common autoimmune disease of which the underlying molecular mechanisms remain largely unclear. Recently, increasing studies based on microarray data profiles have been carried out to elucidate SLE pathogenesis [18, 19]. However, the results generated from a single cohort study are always limited or inconsistent. Interestingly, the combination of integrated bioinformatics methods and expression profiling techniques may overcome the disadvantages. Thus, in this study, we integrated three cohort profile datasets from different places including the USA, Japan, and Belgium, and deeply analyzed these data by integrated bioinformatics methods. A total of 321 commonly changed DEGs were identified, including 231 upregulated and 90 downregulated genes. The 321 common DEGs were mainly enriched in biological processes related to immune responses and inflammatory responses, including innate immune response, defense response, cytokine-mediated signaling pathway, response to interferon-alpha, and I-kappaB kinase/NF-kappaB signaling, whereas the three most significant cellular components were oxidoreductase complex, AIM2 inflammasome complex, and ubiquitin ligase complex. Furthermore, the DEGs were primarily involved in several signaling pathways associated with immune system, including RIG-I-like receptor signaling pathway and antigen processing and presentation. These results were in accordance with the knowledge that immune abnormalities and inflammatory response contribute to autoimmune diseases, including SLE. One of the significant enriched pathways, the p53 signaling pathway, has been reported to be strongly associated with apoptosis, cell cycle arrest, and DNA repair. The result was consistent with the characterization of SLE—increased apoptotic cell production and aberrant apoptotic cell clearance [20].
As a common environmental factor, infection plays an important role in the process of SLE onset and development. In this research, KEGG pathway enrichment analysis indicated that common DEGs were enriched in infectious diseases, including influenza A and herpes simplex infection. In addition, GO function analysis showed that common DEGs were primarily enriched in interferon signaling pathway and RIG-I-like receptor signaling pathway. Certain studies have confirmed that type I IFN signature was found in SLE and IFN signaling pathways were strongly associated with the pathogenesis and development of SLE [21, 22]. RIG-I-like receptors (RLRs) are cytosolic sensors for virus RNA and the RIG-I-like receptor signaling induces the secretion of type I interferon and proinflammatory cytokines, leading to activation of immune responses [23]. RLR mutations were found in SLE patients and mice with RLR-related gene mutation spontaneously developed lupus-like symptoms [24]. Recently, Wu’s group analyzed the microarray expression profile GSE65391, including 924 SLE samples and 72 healthy controls, and got the same results as we did [25]. Taken together, the results of this study in association with the other previous studies have indicated a basis for the vital role of interferon signaling pathway and RIG-I-like receptor signaling pathway in the pathogenesis and progression of SLE.
In this work, GO function analysis of DEGs demonstrated that the DEGs were mainly enriched in NF-κB signaling pathway and that FBXW11 was enriched in these pathways. NF-κB signaling pathways consist of the classical NF-κB activation pathway and the alternative pathway, and both pathways play an important role in regulating the immune and inflammatory responses [26,27,28,29]. Previous studies have suggested that high plasma levels of the B cell–activating factor (BAFF) that is known to trigger a non-canonical NF-κB signaling pathway were found in both SLE patients and murine lupus models [27, 30]. In addition, recent studies have indicated that inhibiting the NF-κB signaling pathway ameliorates lupus nephritis in lupus-prone MRL/lpr mice and pristane-induced lupus mice [31,32,33]. These results have demonstrated that NF-κB signaling pathways serve a crucial role in the pathogenesis of SLE. FBXW11, one of the WD-40-containing F-box proteins, is a component of E3 ubiquitin ligase complex that promotes signal-dependent ubiquitination of IκBα and activates the NF-κB signaling pathway [34, 35]. In this study, FBXW11 was identified to be upregulated in SLE patients compared with healthy controls. Although the function of FBXW11 in SLE has not been fully elucidated, we assume that FBXW11 may be implicated in the development of SLE via the NF-κB signaling pathway.
FOXO1 is a member of the forkhead box class O (FOXO) family characterized by a conserved DNA binding domain [36]. In mammals, FOXO1 is involved in regulating various cellular processes and functions, including cell proliferation, apoptosis, oxidative stress responses, metabolism, tumor suppression, and immune responses [37,38,39,40]. A previous study has suggested that FOXO1 transcriptional levels were markedly decreased in the peripheral blood mononuclear cells of SLE patients and inversely correlated with the disease activity [41]. Recently, many studies have demonstrated that FOXO1 plays an important role in the proliferation and activation of B lymphocytes [42,43,44]. Moreover, FOXO1 was identified to negatively regulate Th17 differentiation [45]. MicroRNA-873 was found to facilitate Th17 differentiation by targeting FOXO1 and be implicated in the pathogenesis of systemic lupus erythematosus [46]. In this study, FOXO1 was identified to be downregulated in SLE patients. Thus, the results of this study combined with previous studies indicated that FOXO1 may be involved in the pathogenesis of SLE by regulating proliferation and differentiation of lymphocytes.
SMAD7 is a member of inhibitory Smad family with conserved carboxy-terminal MH2 domains and inhibits TGF-β and BMP-induced Smad signaling [47]. A microarray study revealed that the signaling of a TGF-β-dependent pathway induced fibrosis in discoid lupus erythematosus [48]. Additionally, a previous study has indicated that blocking TGF-β signaling by Smad7 overexpression ameliorated progressive renal injury in a mouse model of autoimmune crescentic glomerulonephritis [49]. However, the biological roles of Smad7 in the development of SLE remain largely unknown. In our study, the expression levels of Smad7 were decreased in SLE patients compared with healthy controls, suggesting that Smad7 may be involved in the progression of SLE.
RPL26L1, a member of the ribosomal protein family, shares high sequence similarity with ribosomal protein L26 [50] and has a role in protein synthesis. The present study has determined RPL26L1 to be upregulated in SLE and suggested that RPL26L1 may be closely related with SLE development. But there were still no researches on the link between RPL26L1 and SLE.
Conclusion
In conclusion, integrated bioinformatics analysis of three profile datasets that based on SLE patients and healthy controls was performed and 321 common DEGs were identified. The common DEGs were significantly enriched in various pathways, especially the pathways associated with immune system and inflammatory responses. Furthermore, four hub genes (RPL26L1, FBXW11, FOXO1, and SMAD7) were validated in SLE patients. Collectively, these results definitely point out the molecular basis to better understand the pathogenesis and provide valuable novel markers or targets for the diagnosis and treatment of SLE.
References
Tsokos GC (2011) Systemic lupus erythematosus. N Engl J Med 365:2110–2121. https://doi.org/10.1056/NEJMra1100359
Carter EE, Barr SG, Clarke AE (2016) The global burden of SLE: prevalence, health disparities and socioeconomic impact. Nat Rev Rheumatol 12:605–620. https://doi.org/10.1038/nrrheum.2016.137
Ohl K, Tenbrock K (2015) Regulatory T cells in systemic lupus erythematosus. Eur J Immunol 45:344–355. https://doi.org/10.1002/eji.201344280
Moulton VR, Tsokos GC (2015) T cell signaling abnormalities contribute to aberrant immune cell function and autoimmunity. J Clin Investig 125:2220–2227. https://doi.org/10.1172/JCI78087
Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, Jahnsen FL (2001) Plasmacytoid dendritic cells (natural interferon- alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol 159:237–243
Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW (2003) Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A 100:2610–2615. https://doi.org/10.1073/pnas.0337679100
Le Bon A, Durand V, Kamphuis E, Thompson C, Bulfone-Paus S, Rossmann C et al (2006) Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J Immunol 176:4682–4689
Swanson CL, Wilson TJ, Strauch P, Colonna M, Pelanda R, Torres RM (2010) Type I IFN enhances follicular B cell contribution to the T cell-independent antibody response. J Exp Med 207:1485–1500. https://doi.org/10.1084/jem.20092695
Xu HC, Grusdat M, Pandyra AA, Polz R, Huang J, Sharma P, Deenen R, Köhrer K, Rahbar R, Diefenbach A, Gibbert K, Löhning M, Höcker L, Waibler Z, Häussinger D, Mak TW, Ohashi PS, Lang KS, Lang PA (2014) Type I interferon protects antiviral CD8+ T cells from NK cell cytotoxicity. IMMUNITY. 40:949–960. https://doi.org/10.1016/j.immuni.2014.05.004
Donnelly S, Roake W, Brown S, Young P, Naik H, Wordsworth P, Isenberg DA, Reid KBM, Eggleton P (2006) Impaired recognition of apoptotic neutrophils by the C1q/calreticulin and CD91 pathway in systemic lupus erythematosus. Arthritis Rheum 54:1543–1556. https://doi.org/10.1002/art.21783
Jorch SK, Kubes P (2017) An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med 23:279–287. https://doi.org/10.1038/nm.4294
Hung T, Pratt GA, Sundararaman B, Townsend MJ, Chaivorapol C, Bhangale T, Graham RR, Ortmann W, Criswell LA, Yeo GW, Behrens TW (2015) The Ro60 autoantigen binds endogenous retroelements and regulates inflammatory gene expression. SCIENCE. 350:455–459. https://doi.org/10.1126/science.aac7442
Lee HM, Sugino H, Aoki C, Nishimoto N (2011) Underexpression of mitochondrial-DNA encoded ATP synthesis-related genes and DNA repair genes in systemic lupus erythematosus. Arthritis Res Ther 13:R63. https://doi.org/10.1186/ar3317
Lauwerys BR, Hachulla E, Spertini F, Lazaro E, Jorgensen C, Mariette X, Haelterman E, Grouard-Vogel G, Fanget B, Dhellin O, Vandepapelière P, Houssiau FA (2013) Down-regulation of interferon signature in systemic lupus erythematosus patients by active immunization with interferon alpha-kinoid. Arthritis Rheum 65:447–456. https://doi.org/10.1002/art.37785
Ducreux J, Houssiau FA, Vandepapeliere P, Jorgensen C, Lazaro E, Spertini F et al (2016) Interferon alpha kinoid induces neutralizing anti-interferon alpha antibodies that decrease the expression of interferon-induced and B cell activation associated transcripts: analysis of extended follow-up data from the interferon alpha kinoid phase I/II study. Rheumatology (Oxford) 55:1901–1905. https://doi.org/10.1093/rheumatology/kew262
Xia J, Gill EE, Hancock RE (2015) NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat Protoc 10:823–844. https://doi.org/10.1038/nprot.2015.052
Li J, Wang F, Wang G, Sun Y, Cai J, Liu X, Zhang J, Lu X, Li Y, Chen M, Chen L, Jiang C (2017) Combination epidermal growth factor receptor variant III peptide-pulsed dendritic cell vaccine with miR-326 results in enhanced killing on EGFRvIII-positive cells. Oncotarget. 8:26256–26268. https://doi.org/10.18632/oncotarget.15445
Wu C, Zhao Y, Lin Y, Yang X, Yan M, Min Y, Pan Z, Xia S, Shao Q (2018) Bioinformatics analysis of differentially expressed gene profiles associated with systemic lupus erythematosus. Mol Med Rep 17:3591–3598. https://doi.org/10.3892/mmr.2017.8293
Labonte AC, Kegerreis B, Geraci NS, Bachali P, Madamanchi S, Robl R, Catalina MD, Lipsky PE, Grammer AC (2018) Identification of alterations in macrophage activation associated with disease activity in systemic lupus erythematosus. PLoS One 13:e208132. https://doi.org/10.1371/journal.pone.0208132
Tsokos GC, Lo MS, Costa RP, Sullivan KE (2016) New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol 12:716–730. https://doi.org/10.1038/nrrheum.2016.186
Feng X, Wu H, Grossman JM, Hanvivadhanakul P, FitzGerald JD, Park GS et al (2006) Association of increased interferon-inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus erythematosus. Arthritis Rheum 54:2951–2962. https://doi.org/10.1002/art.22044
Weidenbusch M, Kulkarni OP, Anders HJ (2017) The innate immune system in human systemic lupus erythematosus. Clin Sci (Lond) 131:625–634. https://doi.org/10.1042/CS20160415
Dixit E, Kagan JC (2013) Intracellular pathogen detection by RIG-I-like receptors. Adv Immunol 117:99–125. https://doi.org/10.1016/B978-0-12-410524-9.00004-9
Kato H, Fujita T (2015) RIG-I-like receptors and autoimmune diseases. Curr Opin Immunol 37:40–45. https://doi.org/10.1016/j.coi.2015.10.002
Wu C, Zhao Y, Lin Y, Yang X, Yan M, Min Y, Pan Z, Xia S, Shao Q (2017) Bioinformatics analysis of differentially expressed gene profiles associated with systemic lupus erythematosus. Mol Med Rep. https://doi.org/10.3892/mmr.2017.8293
Brown KD, Claudio E, Siebenlist U (2008) The roles of the classical and alternative nuclear factor-kappaB pathways: potential implications for autoimmunity and rheumatoid arthritis. Arthritis Res Ther 10:212. https://doi.org/10.1186/ar2457
Enzler T, Bonizzi G, Silverman GJ, Otero DC, Widhopf GF, Anzelon-Mills A, Rickert RC, Karin M (2006) Alternative and classical NF-kappa B signaling retain autoreactive B cells in the splenic marginal zone and result in lupus-like disease. Immunity 25:403–415. https://doi.org/10.1016/j.immuni.2006.07.010
Barnes PJ, Karin M (1997) Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med 336:1066–1071. https://doi.org/10.1056/NEJM199704103361506
Baeuerle PA, Baichwal VR (1997) NF-kappa B as a frequent target for immunosuppressive and anti-inflammatory molecules. Adv Immunol 65:111–137
Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, Xu W, Parrish-Novak J, Foster D, Lofton-Day C, Moore M, Littau A, Grossman A, Haugen H, Foley K, Blumberg H, Harrison K, Kindsvogel W, Clegg CH (2000) TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. NATURE. 404:995–999. https://doi.org/10.1038/35010115
You Y, Qin Y, Lin X, Yang F, Li J, Sooranna SR, Pinhu L (2015) Methylprednisolone attenuates lipopolysaccharide-induced Fractalkine expression in kidney of Lupus-prone MRL/lpr mice through the NF-kappaB pathway. BMC Nephrol 16:148. https://doi.org/10.1186/s12882-015-0145-y
Hu Q, Yang C, Wang Q, Zeng H, Qin W (2015) Demethylzeylasteral (T-96) treatment ameliorates mice lupus nephritis accompanied by inhibiting activation of NF-kappaB pathway. PLoS One 10:e133724. https://doi.org/10.1371/journal.pone.0133724
Qu H, Bian W, Xu Y (2014) A novel NF-kappaB inhibitor, DHMEQ, ameliorates pristane-induced lupus in mice. Exp Ther Med 8:100–104. https://doi.org/10.3892/etm.2014.1718
Winston JT, Strack P, Beer-Romero P, Chu CY, Elledge SJ, Harper JW (1999) The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev 13:270–283
Shi P, Zhu S, Lin Y, Liu Y, Liu Y, Chen Z, Shi Y, Qian Y (2011) Persistent stimulation with interleukin-17 desensitizes cells through SCFbeta-TrCP-mediated degradation of Act1. Sci Signal 4:a73. https://doi.org/10.1126/scisignal.2001653
Li Y, Ma Z, Jiang S, Hu W, Li T, Di S et al (2017) A global perspective on FOXO1 in lipid metabolism and lipid-related diseases. Prog Lipid Res 66:42–49. https://doi.org/10.1016/j.plipres.2017.04.002
Zhang X, Tang N, Hadden TJ, Rishi AK (2011) Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta 1813:1978–1986. https://doi.org/10.1016/j.bbamcr.2011.03.010
Martins R, Lithgow GJ, Link W (2016) Long live FOXO: unraveling the role of FOXO proteins in aging and longevity. Aging Cell 15:196–207. https://doi.org/10.1111/acel.12427
Vogt PK, Jiang H, Aoki M (2005) Triple layer control: phosphorylation, acetylation and ubiquitination of FOXO proteins. Cell Cycle 4:908–913. https://doi.org/10.4161/cc.4.7.1796
Malik S, Sadhu S, Elesela S, Pandey RP, Chawla AS, Sharma D, Panda L, Rathore D, Ghosh B, Ahuja V, Awasthi A (2017) Transcription factor Foxo1 is essential for IL-9 induction in T helper cells. Nat Commun 8:815. https://doi.org/10.1038/s41467-017-00674-6
Kuo CC, Lin SC (2007) Altered FOXO1 transcript levels in peripheral blood mononuclear cells of systemic lupus erythematosus and rheumatoid arthritis patients. Mol Med 13:561–566. https://doi.org/10.2119/2007-00021.Kuo
Omori SA, Cato MH, Anzelon-Mills A, Puri KD, Shapiro-Shelef M, Calame K, Rickert RC (2006) Regulation of class-switch recombination and plasma cell differentiation by phosphatidylinositol 3-kinase signaling. Immunity 25:545–557. https://doi.org/10.1016/j.immuni.2006.08.015
Chen J, Yusuf I, Andersen HM, Fruman DA (2006) FOXO transcription factors cooperate with delta EF1 to activate growth suppressive genes in B lymphocytes. J Immunol 176:2711–2721
Yusuf I, Zhu X, Kharas MG, Chen J, Fruman DA (2004) Optimal B-cell proliferation requires phosphoinositide 3-kinase-dependent inactivation of FOXO transcription factors. Blood 104:784–787. https://doi.org/10.1182/blood-2003-09-3071
Laine A, Martin B, Luka M, Mir L, Auffray C, Lucas B et al (2015) Foxo1 is a T cell-intrinsic inhibitor of the RORgammat-Th17 program. J Immunol 195:1791–1803. https://doi.org/10.4049/jimmunol.1500849
Liu L, Liu Y, Yuan M, Xu L, Sun H (2017) Elevated expression of microRNA-873 facilitates Th17 differentiation by targeting forkhead box O1 (Foxo1) in the pathogenesis of systemic lupus erythematosus. Biochem Biophys Res Commun 492:453–460. https://doi.org/10.1016/j.bbrc.2017.08.075
Miyazawa K, Miyazono K. Regulation of TGF-beta family signaling by inhibitory Smads. Cold Spring Harb Perspect Biol 2017;9. doi: https://doi.org/10.1101/cshperspect.a022095
Sole C, Gimenez-Barcons M, Ferrer B, Ordi-Ros J, Cortes-Hernandez J (2016) Microarray study reveals a transforming growth factor-beta-dependent mechanism of fibrosis in discoid lupus erythematosus. Br J Dermatol 175:302–313. https://doi.org/10.1111/bjd.14539
Ka SM, Huang XR, Lan HY, Tsai PY, Yang SM, Shui HA, Chen A (2007) Smad7 gene therapy ameliorates an autoimmune crescentic glomerulonephritis in mice. J Am Soc Nephrol 18:1777–1788. https://doi.org/10.1681/ASN.2006080901
Towers CG, Guarnieri AL, Micalizzi DS, Harrell JC, Gillen AE, Kim J, Wang CA, Oliphant MUJ, Drasin DJ, Guney MA, Kabos P, Sartorius CA, Tan AC, Perou CM, Espinosa JM, Ford HL (2015) The Six1 oncoprotein downregulates p53 via concomitant regulation of RPL26 and microRNA-27a-3p. Nat Commun 6:10077. https://doi.org/10.1038/ncomms10077
Acknowledgments
We gratefully thank many researchers for providing technical assistance and all the patients who were enrolled in the study.
Funding
This work was supported by grants from National Natural Science Foundation of China (No. 81501417, No. 81671623) and Science and Technology Planning Project of Guangdong Province, China (2014B020212024).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Declaration of conflicting interests
The authors declare that there is no conflict of interest.
Ethics approval and consent to participate
The study was approved by the Ethics Committee of The Third Affiliated Hospital of Southern Medical University. Informed consent has been obtained from the patients’ legal guardians.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 27 kb)
Rights and permissions
About this article

Cite this article
Yang, F., Zhai, Z., Luo, X. et al. Bioinformatics identification of key candidate genes and pathways associated with systemic lupus erythematosus. Clin Rheumatol 39, 425–434 (2020). https://doi.org/10.1007/s10067-019-04751-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-019-04751-7