
Abstract
Giant axonal neuropathy (GAN) is an autosomal recessive disease caused by mutations in the GAN gene encoding gigaxonin. Patients develop a progressive sensorimotor neuropathy affecting peripheral nervous system (PNS) and central nervous system (CNS). Methods: In this multicenter observational retrospective study, we recorded French patients with GAN mutations, and 10 patients were identified. Mean age of patients was 9.7 years (2–18), eight patients were female (80%), and all patients met infant developmental milestones and had a family history of consanguinity. Mean age at disease onset was 3.3 years (1–5), and progressive cerebellar ataxia and distal motor weakness were the initial symptoms in all cases. Proximal motor weakness and bulbar symptoms appeared at a mean age of 12 years (8–14), and patients used a wheelchair at a mean age of 16 years (14–18). One patient died at age 18 years from aspiration pneumonia. In all cases, nerve conduction studies showed a mixed demyelinating and axonal sensorimotor neuropathy and MRI showed brain and cerebellum white matter abnormalities. Polyneuropathy and encephalopathy both aggravated during the course of the disease. Patients also showed a variety of associated findings, including curly hair (100% of cases), pes cavus (80%), ophthalmic abnormalities (30%), and scoliosis (30%). Five new GAN mutations were found, including the first synonymous mutation and a large intragenic deletion. Our findings expand the genotypic spectrum of GAN mutations, with relevant implications for molecular analysis of this gene, and confirm that GAN is an age-related progressive neurodegenerative disease involving PNS and CNS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Giant axonal neuropathy (GAN, OMIM #256850), is an autosomal recessive progressive disease affecting the peripheral and central nervous systems (PNS and CNS). “Kinky hair” (tightly curled hair not observed in parents) and long eyelashes are characteristic features of the disease, albeit not observed in all cases [1]. GAN usually presents as a childhood-onset axonal neuropathy, with a majority of patients losing ambulation in their teens and loosing life in their thirties [1]. CNS involvement may include leukoencephalopathy, optic atrophy, nystagmus, cerebellar dysfunction, intellectual disability, and spasticity [1]. Nerve biopsy shows reduced density of nerve fibers and large axons with neurofilament accumulation [1].
The gene causing GAN was identified in the year 2000 and more than 80 different GAN mutations have been described in approximately 100 patients since then [2,3,4,5,6,7,8,9,10,11,12]. GAN encodes the 65-kDa protein gigaxonin, a BTB/Kelch protein with an N-terminal BTB (Bric-a-brac, Tramtrack and Broad) domain, central BACK domain, and 6 Kelch repeats [1]. There is no clustering of mutations, and a majority of patients present with point mutations in the BTB, BACK, and 6 Kelch repeat domains [1]. It was suggested that GAN was an E3 ligase substrate adaptor possibly involved in the regulation of intermediate filaments (IFs) turnover [1]. To date, there is no treatment for GAN.
In this multicenter observational retrospective study, we report the detailed molecular, electrophysiological, brain magnetic resonance imaging (MRI), and clinical characteristics of 10 patients with 5 new GAN mutations, including 1 patient with a large intragenic deletion and 2 patients with a synonymous mutation.
Patients and methods
Protocol approvals, patients’ consents, and registration
Patients
We performed an observational retrospective study including French patients with pathogenic homozygous variations in the GAN gene. Patients were identified from French laboratories sequencing the GAN gene (Lyon, Paris), and data collection was performed in hospitals following the patients. Six pediatricians and neurologists were contacted, and clinical, paraclinical, and genetic data were compiled from medical records by the referring physician. Data gathering was performed between January 2018 and September 2018.
Electrophysiological studies
Electrodiagnostic studies were performed in 7 patients including upper and lower limbs nerve conduction studies (NCS) and electromyography (EMG) using a concentric needle electrode in at least 2 muscles. Studies were achieved using regular equipment and accepted methods, and skin temperature was kept in 32°–34 °C range.
Additional tests
All patients underwent pulmonary function tests (PFT) and standardized biological tests. Brain MRI was performed in 6 patients. Vocal cord mobility was investigated using laryngoscopy in 3 patients with dysphonia. Muscle biopsy was performed in 1 patient and processed with standard methods for histology and histochemistry.
Genetic analysis
The 11 exon and intron junctions of the GAN gene were sequenced by Sanger di-deoxynucleotide method in all patients. Numbering of mutations was done using RefSeq cDNA NM_022489.3. All patients gave informed consent for genetic analysis.
In silico splicing prediction
No cDNA sample was available in family F2, and we used 4 splice site algorithms to predict a potential splicing effect: MaxEntScan (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html), NNSplice (http://www.fruitfly.org/), GeneSplicer (http://www.cbcb.umd.edu/software/GeneSplicer/gene_spl.shtml), and Human Splicing Finder (http://www.umd.be/HSF/) [13,14,15].
Minigene constructs & in vitro splicing test
Minigene constructions for splicing studies were performed as previously described using the (CTAAACAGCCACATATGACGGATGCCTCAGATCTGGG) and (CCCCCCTCGACCATATGGGCAGGATAAATGTGAAGGC) primers [16]. The sequence we analyzed included the end of GAN intron 5 (400 nucleotides), the entire exon 6 (113 nucleotides), and the beginning of intron 6 (243 nucleotides). Human HeLa cells were used for triplicate transfections [16].
Results
Patients
We found 10 patients from 5 distinct families (Table 1). Mean age of patients was 9.7 years (2–18), eight patients were female (80%), and all patients met infant developmental milestones and had a family history of consanguinity (Table 2). Mean age at disease onset was 3.3 years (1–5), and progressive cerebellar ataxia and distal motor weakness were the initial symptoms in all cases (Fig. 1). Proximal motor weakness and bulbar symptoms, i.e., dysphonia and dysphagia, appeared at a mean age of 12 years (8–14), and patients used a wheelchair at a mean age of 16 years (14–18) (Fig. 2). One patient died at age 18 of aspiration pneumonia. Patients also showed an assortment of associated findings, including curly hair (100% of cases), pes cavus (80%), ophthalmic abnormalities (30%), scoliosis (30%), tendon contractures (20%), hirsutism (20%), precocious puberty (10%), vocal cord paralysis (10%), and hearing impairment (10%) (Table 2).

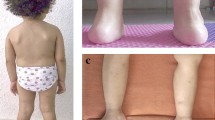
Clinical features of patients with GAN mutations a Patient 3, age 6 years: distal lower limbs atrophy (black arrows). b Patient 10, age 15 years: distal upper limbs atrophy (white arrows). c Patient 2, age 10 years: curly hair

Time of occurrence of the main clinical features Distal motor weakness appeared at a mean age of 2.9 years (1–5, n = 10); cerebellar ataxia at a mean age of 3.3 years (1–5; n = 10); bulbar symptoms at a mean age of 10.4 years (8–14; n = 4); proximal motor weakness at a mean age of 12 years (8–14; n = 3); and ambulation loss at a mean age of 16 years (14–18; n = 2). One patient died at age of 18 years.
Electrophysiological studies
NCS studies showed abnormal upper & lower limb compound muscle action potentials (CMAPs), sensory nerve action potential (SNAPs), and motor and sensory nerve conduction velocities (MSNCVs) in all 7 investigated cases (Supplemental Table 1). EMG demonstrated moderate neuropathic changes in distal lower limb muscles of all patients, and we found no pathological spontaneous activity. In our series, CMAPs, SNAPs, and MSNCVs alterations tend to aggravate with age, with sensory and motor responses progressively disappearing after 9 years of age (Supplemental Table 1). To summarize, our patients had a slowly progressive, symmetrical, predominantly distal, sensorimotor neuropathy with axonal and demyelinating features.
Additional tests
Brain MRI was abnormal in all 6 investigated cases (Fig. 3). Cerebellar abnormalities, including abnormal dentate nuclei and leukopathy, were observed in all cases (Fig. 3). Brain white matter abnormalities were observed in all cases, with optical radiations involved at an early stage, and internal capsule, periventricular and subcortical white matter, thalami, and brainstem involved at later stages (Fig. 3). Taken together, our data demonstrate that our patients had a slowly progressive diffuse leukoencephalopathy involving brain, cerebellum, and brainstem. Laryngoscopy showed vocal cord paralysis in 1 patient with dysphonia. In one case, tibialis anterior muscle biopsy found neurogenic abnormalities.

Brain MRI of patients with GAN mutations Brain MRI of three patients with GAN mutations at age 6 years (1a–d), 7 years (2a–e), and 14 years (3a–b). MRI shows cerebellar abnormalities in all patients at all ages, including abnormal dentate nuclei with a central hyperintense signal surrounded by a hypointense signal observed in T2-FLAIR sequences (1a–b; 2a arrows) and T2-weighted sequences (3a–b; arrows), and cerebellar leukopathy (2b; asterisk). MRI also shows brain white matter abnormalities involving optical radiations at an early stage (1d; asterisk), and involving internal capsule, periventricular and subcortical white matter, thalami, and brainstem at an advanced stage (2c–e; asterisks).
Genetic analysis and in silico splicing prediction
Five new homozygous mutations in the GAN gene were identified in the 5 families reported in our study, including missense mutations in the GAN Kelch domain in the F3 (Glu493Lys) and F4 (Arg458Trp) families (Fig. 4). GAN mutations segregated in families F1, F2, F4, and family DNA was not available in families F3 and F5. In family F1, exons 2 to 9 of GAN could not be amplified by PCR, and CGH array analysis confirmed the homozygous deletion of GAN exons 2–9 (Fig. 4). In family F2, a homozygous synonymous GAN variant c.1086G>A (p.Glu362=) was found close to the end of exon 6 (Fig. 4). In silico splicing analysis predicted an abolition of the donor site of exon 6 (Supplemental Table 2). In family F3, a rare GAN homozygous p.Glu493Lys (c.1477G>A) (rs780427229) variant was identified in exon 9 (Fig. 4). The frequency of this variant in gnomAD database was 0.0018% for non-Finnish Europeans, the affected amino acid was conserved in vertebrates, and bioinformatic tools predicted the variation to be deleterious (PolyPhen-2: probably damaging, score 0.999; SIFT: tolerated, score 0.26) (Supplemental Table 3). In family F4, a GAN homozygous p.Arg458Trp (c.1372A>T) variation was found at the end of exon 8 (Fig. 4). The mutation was absent in the 1000 Genomes, ExAC, and gnomAD databases, affected an amino-acid conserved from zebrafish to humans, and in silico predictions were consistent with a deleterious substitution (SIFT: deleterious, score 0; PolyPhen-2: probably damaging, score 1.000) (Supplemental Table 3). Codon 458 overlaps exons 8 and 9, and in silico analysis predicted a limited impact on the donor site of exon 8 (mean value− 15 %). In family F5, a GAN homozygous p.Ser52Asn (c.155G>A) variation was found in exon 1 (Fig. 4). The variant was absent in the 1000 Genomes, ExAC, and gnomAD databases, and affected an amino-acid conserved from zebrafish to humans (Supplemental Table 3). To note, a mutation on the same codon (p.Ser52Gly) was previously reported in the patient with GAN (Bomont 2000).

GAN mutations reported in this study Schematic representation of GAN cDNA in yellow with functional domains in orange. The 5 new mutations described in this study (S52N, E362=, R458W, E493K, and exons 2–9 deletion) are on top of the figure, and their corresponding exons are delineated with dotted lines. Previously reported missense mutations are in gray (Johnson-Kerner BL et al., Muscle Nerve 2014; 50:467–76; Wang L et al., Muscle Nerve 2014; 50:200–5; Roth LA et al., Neuromuscul Disord 2014; 24 :48–55; Koichihara R et al., Brain Dev 2016; 38:350–3)
In vitro splicing assay analysis
As no cDNA was available in family F2, minigene experiments were performed to investigate the molecular effects of the p.Glu362=variant. Minigene experiments revealed a reduction of the 360 bp normal GAN mRNA and the presence of an alternate 375 bp splicing product (Fig. 5a). Sequencing of the minigene product demonstrated that the first 15 nucleotides of GAN intron 6, which contain an in-frame stop codon, were retained (Fig. 5b). At protein level, retention of this intronic in-frame stop codon provokes the appearance of a premature stop codon at position 366 (Fig. 5b).

In vitro splicing assay for the GAN c.1086G>A (p.Glu362=) mutation a The c.1086C>A mutation provokes a massive reduction of the 360 bp normal GAN mRNA and the appearance of an alternate 375 bp splicing product. b Sequencing of the minigene product demonstrates the retention of the first 15 nucleotides of GAN intron 6 which contain an in-frame stop codon. At protein level, the retention of this intronic in-frame stop codon provokes the appearance of a premature stop codon at position 366
Discussion
In this multicenter observational retrospective study, we identified 10 patients with 5 new GAN mutations, including 1 patient with the first large intragenic GAN deletion (exons 2–9 deletion) and 2 patients with the first GAN synonymous mutation (p.Glu362=).
The phenotypic spectrum observed in our series of 10 patients was homogeneous, with progressive cerebellar ataxia and distal motor weakness appearing at age 1–5 years, later followed by proximal motor weakness and bulbar symptoms appearing at age 8–14 years, and eventually loss of ambulation appearing at age 14–16 years (Table 2, Figs. 1 and 2). Friedreich Ataxia Rating Scale & the Gross Motor Function measure, which have been recently validated to assess motor function in GAN, were not analyzed in our patients [17]. Importantly, all patients in our series met infant developmental milestones, had a family history of consanguinity, and presented with curly hair, the latter being in our series as an excellent clinical marker of the disease. However, not all GAN patients have curly hair, and it has been suggested that straight hair may be correlated with a phenotypically milder disease [1, 9]. More recently, the use of next generation sequencing (NGS), including exome sequencing and targeted gene panel analysis, has shown that GAN mutations can be associated in a few families with a typical intermediate or axonal Charcot-Marie-Tooth phenotype (CMT2) without any additional signs and symptoms [11, 18, 19]. Eventually, the clinical findings we noted are in accordance with other series demonstrating that GAN mutations provoke a progressive sensorimotor neuropathy affecting both PNS and CNS [1].
In our series, brain MRI was abnormal in all 6 investigated cases (Fig. 3). Interestingly, in contrast with cerebellar abnormalities which were observed in all cases at all ages, brain white matter abnormalities progressively spread with age, with optical radiations involved at an early stage, and internal capsule, periventricular and subcortical white matter, thalami, and brainstem involved at later stages (Fig. 3). Similar MRI abnormalities have been observed in several GAN cases, but the course of GAN CNS MRI abnormalities are hard to scrutinize because of the condition scarcity [1, 6]. Interestingly, our results suggest that GAN mutations provoke an age-related progressive leukoencephalopathy initially involving the cerebellum, and latter spreading to the brain hemispheres and brainstem (Fig. 3).
When considering electrodiagnostic studies, our data demonstrate that our patients presented with a slowly progressive, symmetrical, predominantly distal, axonal, and demyelinating sensorimotor neuropathy (Supplemental Table 1). For the most part, these findings are in accordance with other published series [1]. Interestingly, in our series, CMAPs, SNAPs, and MSNCVs alterations tended to aggravate with age, with sensory and motor responses progressively disappearing after 9 years of age, suggesting age-related progressive axon loss (Supplemental Table 1).
In our study, we found 5 new deleterious GAN mutations in 5 families, including the first GAN synonymous mutation (p.Glu362=) and a large GAN large intragenic deletion (exons 2–9 deletion) (Fig. 4, Supplemental Table 2). To clarify the effect of the c.1086G>A (p.Glu362=) synonymous variant, we performed in vitro splicing assays which showed that this variation provokes the appearance of an in-frame cryptic premature stop codon in intron 6 (Fig. 5). The splicing assay confirmed the abolition of the exon 6 donor site predicted by the 4 prediction algorithms (Supplemental Table 3). At protein level, the synonymous p.Glu362= mutation is therefore most likely a frameshift mutation with a premature stop codon (p.Ala363Valfs*3). Overall, these results are in accordance with what has been previously shown in GAN, i.e., the absence of mutation clustering, the predominance of missense point mutations, and the scarcity of intragenic deletions [1, 20, 21].
GAN pathological studies usually show enlarged axons filled with compact, densely bundled IFs [1]. GAN postmortem studies usually show PNS and CNS neurodegeneration, including axon swellings and spheroids in the spinal cord, brainstem, and cerebral cortex (reviewed in [1]). In GAN, a derangement of many IF classes is observed in neurons, astrocytes, Schwann cells, perineurial cells, endothelial cells, muscle fibers, fibroblasts, and lens epithelial cells [22,23,24]. Inclusion content is cell-type dependent and includes neurofilaments (NFs), peripherin, alpha-internexin, vimentin, glial fibrillary acidic protein, nestin, desmin, and keratin [22, 23, 25]. The exact role of GAN protein is unknown, although its role as an E3 ligase substrate adaptor involved in IF turnover regulation is strongly suspected [22, 23, 25]. There is no correlation between GAN protein level and disease severity, suggesting that the amount of gigaxonin needed in a cell has a critical value [22, 23, 25].
There is currently no approved therapy for GAN patients. However, it has been recently shown that an adeno-associated viral (AAV) vector bearing a normal copy of the human GAN transgene (AAV9/JeT-GAN) restored normal IFs configuration in patient fibroblasts and GAN knock-out (KO) mice [26]. Interestingly, intrathecal delivery of AAV9/JeT-GAN in GAN KO mice led to conserved sciatic nerve ultrastructure, lowered IF accumulations in neurons, alleviated rotarod dysfunction, and maintained wild-type PNS and CNS gigaxonin expression [24]. As a result, a phase I trial using viral-mediated GAN gene replacement is underway (https://clinicaltrials.gov/ct2/show/NCT02362438) [26].
Contributorship statement
AEL, JMC, LGM, PA, LK, NB, AV, KB, JP, BF, and PL designed the research, performed the research, and participated in the data collection. Andoni Echaniz-Laguna and Philippe Latour wrote the manuscript. JMC, LGM, PA, LK, NB, AV, KB, JP, and BF revised the manuscript for intellectual content.
References
Johnson-Kerner BL, Roth L, Greene JP, Wichterle H, Sproule DM (2014) Giant axonal neuropathy: an updated perspective on its pathology and pathogenesis. Muscle Nerve 50:467–476
Bomont P, Cavalier L, Blondeau F, Ben Hamida C, Belal S, Tazir M, Demir E, Topaloglu H, Korinthenberg R, Tüysüz B, Landrieu P, Hentati F, Koenig M (2000) The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet 26:370–374
Tazir M, Vallat JM, Bomont P, Zemmouri R, Sindou P, Assami S, Nouioua S, Hammadouche T, Grid D, Koenig M (2002) Genetic heterogeneity in giant axonal neuropathy: an Algerian family not linked to chromosome 16q24.1. Neuromuscul Disord 12:849–852
Bomont P, Ioos C, Yalcinkaya C et al (2003) Identification of seven novel mutations in the GAN gene. Hum Mutat 21:446
Bruno C, Bertini E, Federico A et al (2004) Clinical and molecular findings in patients with giant axonal neuropathy (GAN). Neurology 62:13–16
Demir E, Bomont P, Erdem S et al (2005) Giant axonal neuropathy: clinical and genetic study in six cases. J Neurol Neurosurg Psychiatry 76:825–832
Houlden H, Groves M, Miedzybrodzka Z et al (2007) New mutations, genotype phenotype studies and manifesting carriers in giant axonal neuropathy. J Neurol Neurosurg Psychiatry 78:1267–1270
Tazir M, Nouioua S, Magy L et al (2009) Phenotypic variability in giant axonal neuropathy. Neuromuscul Disord 19:270–274
Roth LA, Johnson-Kerner BL, Marra JD, LaMarca NH, Sproule DM (2014) The absence of curly hair is associated with a milder phenotype in giant axonal neuropathy. Neuromuscul Disord 24:48–55
Koichihara R, Saito T, Ishiyama A et al (2016) A mild case of giant axonal neuropathy without central nervous system manifestation. Brain Dev 38:350–353
Aharoni S, Barwick KE, Straussberg R, Harlalka et al (2016) Novel homozygous missense mutation in GAN associated with Charcot-Marie-Tooth disease type 2 in a large consanguineous family from Israel. BMC Med Genet 17:82
Normendez-Martínez MI, Monterde-Cruz L, Martínez R, Marquez-Harper M, Esquitin-Garduño N, Valdes-Flores M, Casas-Avila L, de Leon-Suarez VP, Romero-Díaz VJ, Hidalgo-Bravo A (2018) Two novel mutations in the GAN gene causing giant axonal neuropathy. World J Pediatr. 14:298–304
Reese MG, Eeckman FH, Kulp D, Haussler D (1997) Improved splice site detection in Genie. J Comput Biol 4:311–323
Dogan RI, Getoor L, Wilbur WJ, Mount SM (2007) SplicePort: an interactive splice-site analysis tool. Nucleic Acids Res 35:W285–W291
Desmet FO, Hamroun D, Lalande M et al (2009) Human splicing finder: an online bio-neurogenetics informatics tool to predict splicing signals. Nucleic Acids Res 37:e67
Crehalet H, Latour P, Bonnet V et al (2010) U1 snRNA mis-binding: a new cause of CMT1B. Neurogenetics 11:13–19
Roth LA, Marra JD, LaMarca NH, Sproule DM (2015) Measuring disease progression in giant axonal neuropathy: implications for clinical trial design. J Child Neurol 30:741–748
Hoebeke C, Bonello-Palot N, Audic F et al (2018) Retrospective study of 75 children with peripheral inherited neuropathy: genotype-phenotype correlations. Arch Pediatr 25:452–458
Bacquet J, Stojkovic T, Boyer A et al (2018) Molecular diagnosis of inherited peripheral neuropathies by targeted next-generation sequencing: molecular spectrum delineation. BMJ Open 8:e021632
Buysse K, Vergult S, Mussche S, Ceuterick-de Groote C, Speleman F, Menten B, Lissens W, van Coster R (2010) Giant axonal neuropathy caused by compound heterozygosity for a maternally inherited microdeletion and a paternal mutation within the GAN gene. Am J Med Genet A 152A:2802–2804
Boizot A, Talmat-Amar Y, Morrogh D et al (2014) The instability of the BTB-KELCH protein gigaxonin causes giant axonal neuropathy and constitutes a new penetrant and specific diagnostic test. Acta Neuropathol Commun 2:47
Johnson-Kerner BL, Ahmad FS, Diaz AG, Greene JP, Gray SJ, Samulski RJ, Chung WK, van Coster R, Maertens P, Noggle SA, Henderson CE, Wichterle H (2015) Intermediate filament protein accumulation in motor neurons derived from giant axonal neuropathy iPSCs rescued by restoration of gigaxonin. Hum Mol Genet 24:1420–1431
Johnson-Kerner BL, Garcia Diaz A, Ekins S, Wichterle H (2015) Kelch Domain of gigaxonin interacts with intermediate filament proteins affected in giant axonal neuropathy. PLoS One 10:e0140157
Armao D, Bouldin TW, Bailey RM et al (2019) Advancing the pathologic phenotype of giant axonal neuropathy: early involvement of the ocular lens. Orphanet J Rare Dis 14:27
Mahammad S, Murthy SN, Didonna A, Grin B, Israeli E, Perrot R, Bomont P, Julien JP, Kuczmarski E, Opal P, Goldman RD (2013) Giant axonal neuropathy-associated gigaxonin mutations impair intermediate filament protein degradation. J Clin Invest 123:1964–1975
Bailey RM, Armao D, Nagabhushan Kalburgi S, Gray SJ (2018) Development of intrathecal AAV9 gene therapy for giant axonal neuropathy. Mol Ther Methods Clin Dev:160–171
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This study was accepted and achieved under the ethical guidelines issued by our institutions for clinical studies. This study was in compliance with the Helsinki Declaration. All patients gave informed consent.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOC 52 kb)
Rights and permissions
About this article

Cite this article
Echaniz-Laguna, A., Cuisset, JM., Guyant-Marechal, L. et al. Giant axonal neuropathy: a multicenter retrospective study with genotypic spectrum expansion. Neurogenetics 21, 29–37 (2020). https://doi.org/10.1007/s10048-019-00596-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-019-00596-z