
Abstract
Parkinson’s disease (PD) is a common neurodegenerative disorder of complex aetiology. Rare, highly penetrant PD-causing mutations and common risk factors of small effect size have been identified in several genes/loci. However, these mutations and risk factors only explain a fraction of the disease burden, suggesting that additional, substantial genetic determinants remain to be found. Genetically isolated populations offer advantages for dissecting the genetic architecture of complex disorders, such as PD. We performed exome sequencing in 100 unrelated PD patients from Sardinia, a genetic isolate. SNPs absent from dbSNP129 and 1000 Genomes, shared by at least five patients, and of functional effects were genotyped in an independent Sardinian case-control sample (n = 500). Variants associated with PD with nominal p value <0.05 and those with odds ratio (OR) ≥3 were validated by Sanger sequencing and typed in a replication sample of 2965 patients and 2678 controls from Italy, Spain, and Portugal. We identified novel moderately rare variants in several genes, including SCAPER, HYDIN, UBE2H, EZR, MMRN2 and OGFOD1 that were specifically present in PD patients or enriched among them, nominating these as novel candidate risk genes for PD, although no variants achieved genome-wide significance after Bonferroni correction. Our results suggest that the genetic bases of PD are highly heterogeneous, with implications for the design of future large-scale exome or whole-genome analyses of this disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) occurs in more than 1 % of people aged 65 years and older [1]. Rare, highly penetrant mutations in different genes have been identified in Mendelian forms of PD [2]. Furthermore, common risk factors of small size effect in several loci have been identified for PD [3]. However, these variants explain small percentages of the disease heritability [4], suggesting that additional genetic determinants of the common forms of PD remain to be detected. The analysis of genetically isolated populations offers important advantages in this endeavour. The individuals belonging to these populations originate from a small number of founders and have therefore a more homogeneous genetic make-up [5, 6]. Because of its geographical, historical and cultural characteristics, the population of Sardinia has a unique genetic background [7–9]. Evidence of strong Sardinian founder mutations has been described in several medical areas, including neurological disorders, such as Wilson’s disease [10] and amyotrophic lateral sclerosis [11]. In this study, we test the hypothesis that a considerable proportion of PD cases from Sardinia is explained by moderately rare variants with moderate–strong effect sizes. These variants, also termed “goldilocks” alleles [12], are not easily detectable by linkage analyses because of incomplete penetrance or by GWAS studies, because of their rarity. Here, we searched for novel variants by whole exome sequencing (WES) in 100 Sardinian PD cases and tested them for association with PD in an enlarged Sardinian case-control cohort. The best scoring variants were followed up in a replication sample of more than 2500 PD patients and 2500 controls from south European populations.
Subjects and methods
Standard protocol approvals and patient consents
The project was approved by the appropriate institutional review boards and written informed consent was obtained from all participating subjects.
Participants
All Sardinian cases and controls included in the study were born and living in Sardinia, and all their four grandparents were also born in the island. These individuals were recruited at two movement disorders units in southern-central areas of Sardinia: the General Hospital “S. Michele” in Cagliari and the “S. Francesco” Hospital in Nuoro. For replication studies, Italian cases and controls were ascertained at the Parkinson Institute, Istituti Clinici di Perfezionamento, Milan, and at the Institute of Molecular Bioimaging and Physiology, Catanzaro; Spanish cases and controls were ascertained at the Neurology Service, Hospital Clínic of Barcelona; additional Spanish controls were obtained from the Spanish National DNA Bank; Portuguese cases and controls were ascertained at the movement disorders outpatients clinic of the Lisbon University Hospital. The demographic and clinical features of the different samples are reported in the Table 1.
All patients fulfilled the United Kingdom Brain Bank Criteria for the clinical diagnosis of PD [13]. The clinical evaluation of parkinsonism was performed using the Hoehn-Yahr Scale [14] and the Unified Parkinson’s Disease Rating Scale (UPDRS) [15]. Patients were classified as sporadic if they reported no other PD cases among their first- and second-degree relatives. Control individuals were spouses of PD cases or subjects examined at the same centres for diseases unrelated to PD; they were eligible for inclusion if they reported a negative family history of PD among first- and second-degree relatives. Moreover, at the time of examination, none of the controls showed any signs or symptoms of PD or other neurodegenerative disorders.
Study design and procedures
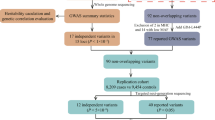
The study design is depicted in Fig. 1. In the discovery sample (100 Sardinian PD), WES was performed at BGI-Shenzhen (see Online Resource Appendix 1 for detailed methods). We aimed at the identification of moderately rare SNP variants (minor allele frequency (MAF) between 0.01 and 0.05) with moderate–strong effect sizes (odds ratio (OR) >2) in the aetiology of PD. Moreover, we hypothesized that a limited number of founder variants could be involved in the disease aetiology in the genetically isolated population of Sardinia. Therefore, out of the SNPs identified in the discovery sample (100 Sardinian PD) we selected for follow-up, the variants fulfilled all the following criteria: (i) absence from both dbSNP129 (http://www.ncbi.nlm.nih.gov/snp/) and 1000 Genomes (http://browser.1000genomes.org/Homo_sapiens/Info/Index, September 2011 data release), (ii) presence in at least five PD patients (regardless of zygosity) and (iii) location within exons and having relevant functional effects (missense, splice-site, nonsense, readthrough) or within 5′- or 3′-UTR. A total number of 5036 SNPs selected in this way (SNP categories are reported in Online Resource Table 1) were typed in the Sardinian case-control cohort (242 PD and 258 controls) by a custom target capturing and next-generation sequencing protocol (refer to Online Resource Appendix 2 for technical specifications). Association of each of these variants with PD status was tested with Fisher’s exact test implemented in PLINK/SEQ (version 0.08) and considering together the entire Sardinian sample (discovery and case-control, total of 600 individuals). From the resulting lists, we prioritized the autosomal variants showing positive association signals (OR >1) with nominal p values <0.05 and the variants with OR ≥3 regardless of the nominal p values. This process yielded 155 variants, which were tested by the Sanger methods in order to remove false positives. None of these 155 SNPs were located in known PD-causing genes. Lastly, the variants confirmed by Sanger sequencing were genotyped in the replication cohorts (5643 individuals) by TaqMan allelic discrimination Assay-by-Design, using an ABI PRISM 7900HT Sequence Detection System and the SDS analysis software, ver.2.4 (Applied Biosystems, Foster City, CA, USA). Final association tests were performed in the entire study sample using Pearson’s chi-square statistic (two-sided asymptotic significance).

Overview of the study design
Duplex real-time quantitative PCR (RT-qPCR) assays were performed using KAPA SYBR FAST qPCR Kit, and a Bio-Rad CFX96 Real-Time System. Data were analysed with CFX Manager software V3.0 (Bio-Rad). Two assays were performed (each in triplicate) targeting different regions of the SNCG gene (exon 1 and exon 5). The RBM and SEL genes were used as reference. qPCR primers and conditions are reported in Online Resource Table 8.
Copy number analysis of the regions flanking the six top candidate variants was performed using Illumina (San Diego, CA, USA) HumanHap300v2 BeadChip array data (318,237 SNPs at a median distance of 5 kb) in the appropriate patients. Data were analysed using Nexus Copy Number, Discovery Edition, version 7.5.2 (Bio-Discovery, El Segundo, CA, USA).
Variants in known PD-causing genes (listed in Online Resource Table 2) were also selected from the discovery samples (100 Sardinian PD) if they were (i) absent from dbSNP129, 1000 Genomes and Exome Variant Server (EVS) or present in EVS with a MAF<0.005; (ii) carried by at least two patients regardless of zygosity; and (iii) of functional effects or located in 5′- and 3′-UTR regions. Six SNPs fulfilled these criteria (Online Resource Table 3), and they all have been confirmed by Sanger sequencing. Among these, two were located in GBA. The genomic regions flanking these GBA variants were amplified in large fragments (to avoid amplification of the neighbouring GBA pseudogene) and subsequently Sanger sequenced in the entire Sardinian case-control cohort (primers and PCR conditions are reported in Online Resource Table 4). The remaining four variants were genotyped by TaqMan allelic discrimination Assay-by-Design in the Sardinian cohort, and those enriched among Sardinian patients were typed in the south European case-control cohorts (Online Resource Table 5). Functional predictions were obtained with PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), sorting intolerant from tolerant (SIFT) (http://sift.jcvi.org/www/SIFT_enst_submit.html) and Grantham Score. For the analysis of the evolutionary conservation, the phastCons (http://genome.ucsc.edu/goldenPath/help/phastCons.html) and GERP (http://mendel.stanford.edu/SidowLab/downloads/gerp/) programs were used. Phosphorylation sites were predicted using the NetPhos 2.0 Server (http://www.cbs.dtu.dk/services/NetPhos/). Fisher’s exact test and Pearson’s chi-square were performed with IBM® SPSS® Statistics (Version 21, release 21.0.0.1). Power calculations were performed with Power for Genetic Association Analyses [16]. The identified variants were submitted to Leiden Open Variation Database (LOVD) v.3.0 (http://www.lovd.nl/3.0/home).
Results
The WES performed in the 100 Sardinian PD cases achieved an average depth of 33.5×, with an overall coverage of 96.33 % of the target region, and 54.58 % of the target covered at least 20 times. A total of 408,601 different SNPs were detected in the 100 Sardinian PD, with an average number of 61,121 SNPs per exome. Among these, 5036 SNPs fulfilled our criteria of absence from both dbSNP129 and 1000 Genomes (September 2011 data release), presence in at least five unrelated PD patients and functional effects for the encoded proteins (missense, splice-site, nonsense, readthrough) or located in the 5′- and 3′-UTR of the transcripts. These 5036 SNPs were included in a custom target capturing system and typed by next-generation sequencing in the Sardinian case-control cohort (500 subjects). The target region was covered at an average depth of 58.5× with 91.97 % covered at least 20 times and almost a full coverage of the targeted bases (99.8 %). Using this method, 4587 out of the 5036 SNPs were successfully typed. Furthermore, due to later updates in the genome reference sequences, additional variants were re-annotated as intergenic and these were removed from our list, leaving a final number of 3881 SNPs. Then, we prioritized the autosomal variants showing association with PD at nominal p value <0.05 and OR >1 and the variants with OR ≥3, regardless of the nominal p values. This process yielded 155 variants, none of which located in known PD-causing genes. Among these, twenty-six variants were validated by PCR and Sanger sequencing, and they are described in Table 2 (PCR primers and protocols are reported in Online Resource Table 6 and Online Resource Table 7). The remaining SNPs were either located in highly repetitive genomic regions or blasting yielded multiple genomic locations, or PCR amplification was unreliable or genotyping yielded inconsistent results. The 26 validated SNPs were then genotyped by Taqman assays in our replication cohorts from southern Europe. We then performed final association analyses in the entire study sample (Online Resource Table 5). Based on our final number of 3881 SNPs selected from the discovery list of variants, the p value requested to declare significant association after Bonferroni correction for multiple testing was 1.28 × 10−5. In the final association analyses, no variants achieved this level of study-wise significance. Furthermore, none of the above-mentioned 26 variants lie within genome-wide association loci for PD (1-Mb window region from the associated SNP) [17].
The allelic frequencies of the six best scoring variants are reported in Table 3.
Copy number analysis of the regions flanking these six variants did not reveal evidence for large aberrations in linkage disequilibrium with our SNP variants (data not shown). In particular, we found no evidence for copy number variants at the SNCG locus.
Concerning the known PD-causing genes, within the discovery samples (100 Sardinian PD), six variants in GBA (n = 2), PARK2 (n = 1), PLA2G6 (n = 1), VPS35 (n = 1) and DNAJC6 (n = 1) genes fulfilled our selection criteria (Online Resource Table 3) and were all validated by Sanger sequencing. We identified the R131C and N370S variants in GBA, with the latter approaching significance for association with PD within the Sardinian case-control cohort (p value 0.07, Fisher’s exact test). The PARK2 R402C and PLA2G6 D31N variants were enriched in the Sardinian PD patients with OR of 2.8 and 1.6, respectively (Online Resource Table 3) but not in the south European case-control cohorts (Online Resource Table 5). Last, the VPS35 and DNAJC6 variants turned out to be equally present among cases and controls within the Sardinian cohort, and they were therefore not followed up.
Discussion
In this study, we tested the hypothesis that moderately rare variants (MAF between 0.01 and 0.05) with moderate–strong effect sizes (OR >2) could underlie a substantial proportions of the PD cases in the genetically isolated population of Sardinia. Because of their intermediate frequencies and effect sizes, this type of variants, also called “goldilocks” alleles, are usually out of reach in traditional linkage and GWAS studies, and they have not previously been systematically addressed on an exome-wide scale in PD.
The case of the LRRK2 and GBA genes provide proofs of principle for the existence of goldilocks alleles as genetic determinants of the common forms of PD. A single variant in the LRRK2 gene (G2019S) is a strong risk factor for PD with odds ratios ∼8–9 and frequencies of ∼30–40 % and ∼15–20 % among PD cases from the Northern-African Arabs and the Ashkenazy Jews population, respectively, while it is rarely observed among controls in those populations (1–2 %) [18]. Moreover, several heterozygous rare GBA variants are strong risk factors (OR >5) for PD in several populations, with one missense variant (N370S) being very frequent among PD in the Ashkenazy Jews population [19].
The analysis of the known PD-causing genes did not reveal any variants with a major role in the Sardinian population. Of note, we identified two GBA variants, N370S and R131C, although the frequencies here are lower compared to other populations [19]. N370S was detected with a minor allele frequency of 1.9 % among Sardinian patients versus 0.7 % among controls (p value 0.07), yielding a lower effect size (OR = 2.5) than earlier reported [20]. In keeping with our results, R131C was shown to be less frequent worldwide, and it was observed by us only in Sardinian patients with a MAF of 0.4 %, compatible with a Mendelian effect. Taken together, these two GBA variants are present in 4.4 % of Parkinson’s disease patients from this genetic isolate.
Identifying PD-associated goldilocks alleles in novel genes was the main goal of our study. Our calculations show that we had adequate power (95 %) to detect the above-mentioned types of moderately rare variants across a range of relative risks between 2 and 10 (Fig. 2). However, the results suggest that none of the inspected variants explains large or moderate percentages of PD cases in the Sardinian population.

Study power calculations. A study sample of 342 cases and 258 controls has 95 % power to detect disease risk variants with moderately rare allelic frequencies (MAF) across a range of relative risks (RR). Power is computed for the detection of association at p values <0.05 after Bonferroni correction for the total n of variants tested in this study (nominal required p value <1.28 × 10−05). All calculations were performed with PGA software (power calculator for case-control genetic association analysis) [16]
These results are compatible with a high degree of genetic or allelic heterogeneity (or both) in the aetiological landscape for PD, even in a genetically isolated population. In this regard, much larger samples of PD cases and controls might be necessary in order to achieve adequate power in the future large-scale exome or whole-genome studies of this disease. Another possibility is to perform gene-centric analyses, and this approach might lead to better results in PD studies [21]. Moreover, sequence-based replication might be more powerful than variant-based replication for both small- and large-scale studies [22].
We acknowledge that this study had limitations. First, in the discovery phase our WES was performed at an average depth of 33.5-fold, and we selected variants shared by at least five PD patients. These facts could have decreased the study sensitivity. However, we reasoned that this design was adequate and cost-effective because we were interested in novel variants present in substantial percentages of PD cases in this genetically homogeneous population. Second, our strategy did not address indels or any variants located in deep intronic or intergenic regulatory elements. Nevertheless, although no variants achieved the level of significance required after stringent Bonferroni correction for multiple testing, our effort nominates a number of interesting candidate variants which are worth following-up in future studies. Particularly, six top variants in SCAPER, HYDIN, UBE2H, EZR, MMRN2 and OGFOD1 might have not reached the significant threshold because they are specific to the Sardinian population but extremely rare in other, outbred populations. Although not significant, they represent novel candidate risk genes for PD, being specifically present in PD patients or enriched among them. The lowest p value (2.77 × 10−03) was obtained for a synonymous substitution in position +1 from a splice site in SCAPER (MIM 611611, Genbank NM_020843.2 and NP_065894.2). This variant is present in 26 Sardinian PD cases and in four Sardinian controls. This gene is ubiquitously expressed in the body including the central nervous system, and it encodes a S-phase cyclin A-associated protein, primarily localized to the endoplasmic reticulum and recently shown to be involved in the progression into S-phase [23]. The second lowest p value (2.92 × 10−03) corresponds to a variant in HYDIN (MIM 610812, NM_001270974.1, NP_001257903.1), leading to a missense change of a glutamate into lysine in the encoded protein. This variant is present in nine Sardinian PD cases and absent among Sardinian controls. This variant has been identified in one PD patient from Milano while it is absent from all control groups and from public databases, and it was scored as “probably damaging” by PolyPhen-2. Although rare, this variant appears to be present only in PD cases, and it could be a genuine determinant of PD. One copy of HYDIN is located on chromosome 16q22.2, and homozygous inactivating mutations therein cause hydrocephalus in mice [24] and primary ciliary dyskinesia without randomization of left-right body asymmetry in humans [25]. However, a paralog gene exists on chromosome 1q21.1 (HYDIN2), which is exclusively expressed in the brain [26]. CNVs at this locus have been associated with micro- and macrocephaly, developmental delay and a wide range of behavioural abnormalities [27]. A variant in UBE2H (MIM 601082, NM_003344.3, NP_003335.1) achieved a p value of 3.59 × 10−03. According to the earlier versions of the reference databases used during the discovery phase of this study, this variant was annotated as a missense; due to this fact, it was included in the replication stage, even if it ultimately turned out to be an intronic variant. This variant is present in 12 Sardinian PD cases and in two Sardinian controls. No carriers are present among the southern European controls or in the public databases. Follow-up studies were possible for two familial Sardinian cases, and co-segregation was observed in one family (the variant was carried by a mother-daughter pair affected by PD). We also detected three additional carriers in the Italian sample from Milano: two with sporadic PD, originating from Central Italy and Sicily and one who is in fact Sardinian and reported a deceased brother affected by PD. UBE2H encodes a member of the E2 ubiquitin-conjugating enzyme family, which catalyse the covalent attachment of ubiquitin to target proteins [28]. Of note, two genes causing Mendelian forms of early-onset PD encode proteins involved in ubiquitination pathways: parkin (PARK2, MIM 602544) [29] and FBXO7 (MIM 605648) [30, 31]. The variant in EZR (MIM 123900, NM_003379.4, NP_003370.2) reached a p value of 6.46 × 10−3 and is predicted to lead to a missense change (phenylalanine into tyrosine). This variant is present in 16 Sardinian PD cases and in four Sardinian controls; it is not present in public databases and is predicted to be damaging by PolyPhen and SIFT. Within the replication samples, two additional Italian PD patients carried this variant, and they turned out to be both of Sardinian origins. EZR encodes ezrin, a member of the Ezrin/Radixin/Moesin (ERM) family. These proteins link the actin cytoskeleton and cell membrane components and are involved in the membrane dynamics during cell adhesion, traffic, signalling and growth [32]. Remarkably, recent studies have identified these ERM proteins as possible physiological substrates of the kinase activity of LRRK2 [33, 34], the protein-product of the gene causing the most common, known form of Mendelian PD (MIM 609007). Furthermore, the ERM proteins and F-actin are downstream targets of LRRK2 during neuronal morphogenesis [34]. Thus, there is compelling evidence that the ezrin variant identified in our study is also a rare but genuine PD-related variant. The variant in MMRN2 (MIM 608925, NM_024756.2, NP_079032.2) is present in nine Sardinian PD cases and absent among Sardinian controls, and achieved a p value of 1.12 × 10−02. MMRN2 encodes an extracellular matrix glycoprotein belonging to the elastin microfibril interface-located (EMILIN) protein family, known to interfere with tumour angiogenesis and growth. Of note, one of the three members of the human synuclein gene family, γ-synuclein (SNCG, MIM 602998, NM_003087.2, NP_003078.2), lies 0.8 kb downstream of MMRN2. The SNCG product, γ-synuclein, is highly homologous to α-synuclein, which is mutated in rare forms of Mendelian PD. Currently, misfolding, aggregation and cell-to-cell prion-like spread of α-synuclein are central themes in the pathogenesis of PD in general [35]. There is also evidence from studies in transgenic mice for a direct involvement of γ-synuclein in the neurodegenerative process [36]. An association between the SNCG locus and diffuse Lewy body disease was also reported [37]. Interestingly, the SNCA and SNCG genes are evolutionary more closely related than SNCB on the basis of their genomic structures [37]. To explore whether our MMRN2 SNP was in linkage disequilibrium (LD) with a different rare variant in SNCG, we sequenced the entire coding region and we carried out copy number analysis of this latter gene (PCR/qPCR primers and protocols are reported in Online Resource Table 8). Only known common SNVs were detected, and copy number variants were not identified. Still, we cannot exclude the presence of a genetic defect in the regulatory elements that could affect the SNCG expression. Interestingly, a similar genomic organization is present on chromosome 6 where the SNCA (MIM 163890) and MMRN1 (MIM 601456) loci are located. This suggests that these two clusters of paralogs (MMRN1 & SNCA on chromosome 14 and MMRN2 & SNCG on chromosome 6) resulted from a large duplication event during evolution [38]. This genomic conservation also suggests the possibility of shared regulatory elements, which could modulate the gene expression.
Last, a 5′-UTR variant in OGFOD1 (eight nucleotides before the ATG-translation initiation codon, NM_018233.3 and NP_060703.3) reached a p value of 1.43 × 10−02. This variant is present in 11 Sardinian PD cases and two Sardinian controls, and it also seems specific for this population. The variant is absent in public databases, and we detected no additional carriers among the southern European samples. OGFOD1 is expressed at substantial levels in the brain and encodes a member of the 2-OG-Fe(II) dioxygenase family, with a role in the cellular survival after ischemia [39]. Moreover, the OGFOD1 protein is a component of the stress granules (cytoplasmic ribonucleoprotein structures composed of repressed translation complexes), implicated in the regulation of translation under cellular stress [40]. There is a recent compelling evidence linking stress granules with misfolding and prion-like propagation of critical proteins implicated in the pathogenesis of neurodegenerative disorders [41, 42]. Thus, OGFOD1 is another interesting candidate for follow-up studies in PD.
In conclusion, this study represents, to our knowledge, the first attempt to identify moderately rare variants of moderate effect size for PD at exome-wide level. Although none of the inspected variants reached the required level of significance after Bonferroni correction for multiple testing, we report several interesting variants only present in PD patients or enriched among them, pointing to novel putative genetic determinants of PD with moderate/strong effect size. We acknowledge that these variants might not be pathogenic themselves but instead be in LD with other deleterious variants in the same or in neighbouring genes, which were missed by the study techniques, and further studies are therefore warranted. The results of our study suggest that with regard to the inspected exome target region, the genetic bases of PD are highly heterogeneous, with important implications for the design of the future exome or whole-genome studies of this common neurodegenerative disease.
References
Wirdefeldt K, Adami HO, Cole P, Trichopoulos D, Mandel J (2011) Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur J Epidemiol 26(Suppl 1):S1–S58
Bonifati V (2014) Genetics of Parkinson’s disease—state of the art, 2013. Parkinsonism Relat Disord 20(Suppl 1):S23–S28
Singleton AB, Farrer MJ, Bonifati V (2013) The genetics of Parkinson’s disease: progress and therapeutic implications. Mov Disord 28:14–23
Keller MF, Saad M, Bras J, Bettella F, Nicolaou N, Simon-Sanchez J, Mittag F, Buchel F, Sharma M, Gibbs JR, Schulte C, Moskvina V, Durr A, Holmans P, Kilarski LL, Guerreiro R, Hernandez DG, Brice A, Ylikotila P, Stefansson H, Majamaa K, Morris HR, Williams N, Gasser T, Heutink P, Wood NW, Hardy J, Martinez M, Singleton AB, Nalls MA, International Parkinson’s Disease Genomics C, Wellcome Trust Case Control C (2012) Using genome-wide complex trait analysis to quantify ‘missing heritability’ in Parkinson’s disease. Hum Mol Genet 21:4996–5009
Peltonen L, Palotie A, Lange K (2000) Use of population isolates for mapping complex traits. Nat Rev Genet 1:182–190
Wang SR, Agarwala V, Flannick J, Chiang CW, Altshuler D, Go TDC, Hirschhorn JN (2014) Simulation of finnish population history, guided by empirical genetic data, to assess power of rare-variant tests in Finland. Am J Hum Genet 94:710–720
Caló CM, Melis A, Vona G, Piras IS (2008) Sardinian population (Italy): a genetic review. Int J Mod Anthropol 1:1–121
Cavalli-Sforza LL, Piazza A, Menozzi P, Mountain J (1988) Reconstruction of human evolution: bringing together genetic, archaeological, and linguistic data. Proc Natl Acad Sci U S A 85:6002–6006
Contu D, Morelli L, Santoni F, Foster JW, Francalacci P, Cucca F (2008) Y-chromosome based evidence for pre-neolithic origin of the genetically homogeneous but diverse Sardinian population: inference for association scans. PLoS One 3:e1430
Loudianos G, Dessi V, Lovicu M, Angius A, Figus A, Lilliu F, De Virgiliis S, Nurchi AM, Deplano A, Moi P, Pirastu M, Cao A (1999) Molecular characterization of wilson disease in the Sardinian population—evidence of a founder effect. Hum Mutat 14:294–303
Chio A, Borghero G, Pugliatti M, Ticca A, Calvo A, Moglia C, Mutani R, Brunetti M, Ossola I, Marrosu MG, Murru MR, Floris G, Cannas A, Parish LD, Cossu P, Abramzon Y, Johnson JO, Nalls MA, Arepalli S, Chong S, Hernandez DG, Traynor BJ, Restagno G, Italian Amyotrophic Lateral Sclerosis Genetic C (2011) Large proportion of amyotrophic lateral sclerosis cases in Sardinia due to a single founder mutation of the TARDBP gene. Arch Neurol 68:594–598
Price AL, Kryukov GV, de Bakker PI, Purcell SM, Staples J, Wei LJ, Sunyaev SR (2010) Pooled association tests for rare variants in exon-resequencing studies. Am J Hum Genet 86:832–838
Hughes AJ, Daniel SE, Kilford L, Lees AJ (1992) Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55:181–184
Hoehn MM, Yahr MD (1967) Parkinsonism: onset, progression and mortality. Neurology 17:427–442
Fahn S, Elton RL, Members of the UPDRS Development Committee (1987) Unified Parkinson’s disease rating scale. Recent developments in Parkinson’s Disease. Macmillan, New York, pp 153–163
Menashe I, Rosenberg PS, Chen BE (2008) PGA: power calculator for case-control genetic association analyses. BMC Genet 9:36
Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, Schulte C, Keller MF, Arepalli S, Letson C, Edsall C, Stefansson H, Liu X, Pliner H, Lee JH, Cheng R, Ikram MA, Ioannidis JP, Hadjigeorgiou GM, Bis JC, Martinez M, Perlmutter JS, Goate A, Marder K, Fiske B, Sutherland M, Xiromerisiou G, Myers RH, Clark LN, Stefansson K, Hardy JA, Heutink P, Chen H, Wood NW, Houlden H, Payami H, Brice A, Scott WK, Gasser T, Bertram L, Eriksson N, Foroud T, Singleton AB, International Parkinson’s Disease Genomics C, Parkinson’s Study Group Parkinson’s Research: The Organized GI, andMe, GenePd, NeuroGenetics Research C, Hussman Institute of Human G, Ashkenazi Jewish Dataset I, Cohorts for H, Aging Research in Genetic E, North American Brain Expression C, United Kingdom Brain Expression C, Greek Parkinson’s Disease C, Alzheimer Genetic Analysis G (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 46:989–993
Correia Guedes L, Ferreira JJ, Rosa MM, Coelho M, Bonifati V, Sampaio C (2010) Worldwide frequency of G2019S LRRK2 mutation in Parkinson’s disease: a systematic review. Parkinsonism Relat Disord 16:237–242
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, De Marco EV, Durr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, Gasser T, Gershoni-Baruch R, Giladi N, Griffith A, Gurevich T, Januario C, Kropp P, Lang AE, Lee-Chen GJ, Lesage S, Marder K, Mata IF, Mirelman A, Mitsui J, Mizuta I, Nicoletti G, Oliveira C, Ottman R, Orr-Urtreger A, Pereira LV, Quattrone A, Rogaeva E, Rolfs A, Rosenbaum H, Rozenberg R, Samii A, Samaddar T, Schulte C, Sharma M, Singleton A, Spitz M, Tan EK, Tayebi N, Toda T, Troiano AR, Tsuji S, Wittstock M, Wolfsberg TG, Wu YR, Zabetian CP, Zhao Y, Ziegler SG (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361:1651–1661
Sidransky E, Lopez G (2012) The link between the GBA gene and parkinsonism. Lancet Neurol 11:986–998
Kiezun A, Garimella K, Do R, Stitziel NO, Neale BM, McLaren PJ, Gupta N, Sklar P, Sullivan PF, Moran JL, Hultman CM, Lichtenstein P, Magnusson P, Lehner T, Shugart YY, Price AL, de Bakker PI, Purcell SM, Sunyaev SR (2012) Exome sequencing and the genetic basis of complex traits. Nat Genet 44:623–630
Liu DJ, Leal SM (2010) Replication strategies for rare variant complex trait association studies via next-generation sequencing. Am J Hum Genet 87:790–801
Tsang WY, Wang L, Chen Z, Sanchez I, Dynlacht BD (2007) SCAPER, a novel cyclin A-interacting protein that regulates cell cycle progression. J Cell Biol 178:621–633
Davy BE, Robinson ML (2003) Congenital hydrocephalus in hy3 mice is caused by a frameshift mutation in Hydin, a large novel gene. Hum Mol Genet 12:1163–1170
Olbrich H, Schmidts M, Werner C, Onoufriadis A, Loges NT, Raidt J, Banki NF, Shoemark A, Burgoyne T, Al Turki S, Hurles ME, Consortium UK, Kohler G, Schroeder J, Nurnberg G, Nurnberg P, Chung EM, Reinhardt R, Marthin JK, Nielsen KG, Mitchison HM, Omran H (2012) Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry. Am J Hum Genet 91:672–684
Doggett NA, Xie G, Meincke LJ, Sutherland RD, Mundt MO, Berbari NS, Davy BE, Robinson ML, Rudd MK, Weber JL, Stallings RL, Han C (2006) A 360-kb interchromosomal duplication of the human HYDIN locus. Genomics 88:762–771
Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T, Lalani SR, Graham B, Lee B, Shinawi M, Shen J, Kang SH, Pursley A, Lotze T, Kennedy G, Lansky-Shafer S, Weaver C, Roeder ER, Grebe TA, Arnold GL, Hutchison T, Reimschisel T, Amato S, Geragthy MT, Innis JW, Obersztyn E, Nowakowska B, Rosengren SS, Bader PI, Grange DK, Naqvi S, Garnica AD, Bernes SM, Fong CT, Summers A, Walters WD, Lupski JR, Stankiewicz P, Cheung SW, Patel A (2008) Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet 40:1466–1471
Kaiser P, Seufert W, Hofferer L, Kofler B, Sachsenmaier C, Herzog H, Jentsch S, Schweiger M, Schneider R (1994) A human ubiquitin-conjugating enzyme homologous to yeast UBC8. J Biol Chem 269:8797–8802
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608
Shojaee S, Sina F, Banihosseini SS, Kazemi MH, Kalhor R, Shahidi GA, Fakhrai-Rad H, Ronaghi M, Elahi E (2008) Genome-wide linkage analysis of a Parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am J Hum Genet 82:1375–1384
Di Fonzo A, Dekker MC, Montagna P, Baruzzi A, Yonova EH, Correia Guedes L, Szczerbinska A, Zhao T, Dubbel-Hulsman LO, Wouters CH, de Graaff E, Oyen WJ, Simons EJ, Breedveld GJ, Oostra BA, Horstink MW, Bonifati V (2009) FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology 72:240–245
McClatchey AI, Fehon RG (2009) Merlin and the ERM proteins—regulators of receptor distribution and signaling at the cell cortex. Trends Cell Biol 19:198–206
Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, Alessi DR (2007) LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson’s disease mutants affect kinase activity. Biochem J 405:307–317
Parisiadou L, Xie C, Cho HJ, Lin X, Gu XL, Long CX, Lobbestael E, Baekelandt V, Taymans JM, Sun L, Cai H (2009) Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J Neurosci 29:13971–13980
Olanow CW, Brundin P (2013) Parkinson’s disease and alpha synuclein: is Parkinson’s disease a prion-like disorder? Mov Disord 28:31–40
Ninkina N, Peters O, Millership S, Salem H, van der Putten H, Buchman VL (2009) Gamma-synucleinopathy: neurodegeneration associated with overexpression of the mouse protein. Hum Mol Genet 18:1779–1794
Nishioka K, Wider C, Vilarino-Guell C, Soto-Ortolaza AI, Lincoln SJ, Kachergus JM, Jasinska-Myga B, Ross OA, Rajput A, Robinson CA, Ferman TJ, Wszolek ZK, Dickson DW, Farrer MJ (2010) Association of alpha-, beta-, and gamma-Synuclein with diffuse Lewy body disease. Arch Neurol 67:970–975
Specht CG, Schoepfer R (2004) Deletion of multimerin-1 in alpha-synuclein-deficient mice. Genomics 83:1176–1178
Saito K, Adachi N, Koyama H, Matsushita M (2010) OGFOD1, a member of the 2-oxoglutarate and iron dependent dioxygenase family, functions in ischemic signaling. FEBS Lett 584:3340–3347
Wehner KA, Schutz S, Sarnow P (2010) OGFOD1, a novel modulator of eukaryotic translation initiation factor 2alpha phosphorylation and the cellular response to stress. Mol Cell Biol 30:2006–2016
Wolozin B (2012) Regulated protein aggregation: stress granules and neurodegeneration. Mol Neurodegener 7:56
Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP (2013) Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495:467–473
Acknowledgments
This work was supported by BGI-Shenzhen, China, and by grants from the “Stichting ParkinsonFonds”—The Netherlands and the Netherlands Organization for Scientific Research (NWO, VIDI grant, project n. 91786395) to V.B. The samples from Milan, Italy, were obtained from the “Parkinson Institute Biobank” (http://www.parkinsonbiobank.com), member of the Telethon Network of Genetic Biobanks (project n. GTB12001) funded by TELETHON Italy and supported by the “Fondazione Grigioni per il Morbo di Parkinson”.
Ethical standards
All experiments performed in this study comply with the current laws of the countries in which they were performed.
Conflict of interest
MQ, XY, GC, SO, VMS, GJB, LO, JH, NX, JG, VR, DM, CM, MJM, PT, RS, FV, MG, GP, ME, AQ, GA, BAO, MM and JW reports no conflict of interest. LCG reports personal fees from Centro Hospitalar Lisboa Norte. JF reports personal fees from GlaxoSmithKline, Novartis, TEVA, Lundbeck, Solvay, Abbott, BIAL, Merck-Serono, Merz, Ipsen, Grunenthal, Merck Sharp and Dohme, Allergan, Centro Hospitalar Lisboa Norte, Faculdade de Medicina de Lisboa, Fundacao MSD (Portugal) and European Huntington Disease Network. SG reports grants from Italian Telethon Foundation. ET received honoraria for consultancy from Novartis, TEVA, Boehringer Ingelheim, UCB, Solvay and Lundbeck, and he received funding for research from the Spaniard Network for Research on Neurodegenerative Disorders (CIBERNED)-Instituto Carlos III (ISCIII), The Michael J. Fox Foundation for Parkinson’s Research (MJFF) and Fondo de Investigaciones Sanitarias de la Seguridad Social (FISS). VB reports personal compensation for serving as Associate Editor of Parkinsonism & Related Disorders and Section Editor of Current Neurology and Neuroscience Reports, honoraria from the International Parkinson and Movement Disorder Society and Elsevier Ltd and research support from the Erasmus MC, Rotterdam, the Stichting ParkinsonFonds (The Netherlands) and The Netherlands Organization for Scientific Research (NWO).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Marialuisa Quadri, Xu Yang, Giovanni Cossu and Simone Olgiati contributed equally to this work and should be considered as joint first authors.
Jun Wang and Vincenzo Bonifati contributed equally to this work and should be considered as joint senior author.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 115 kb)
Rights and permissions
About this article

Cite this article
Quadri, M., Yang, X., Cossu, G. et al. An exome study of Parkinson’s disease in Sardinia, a Mediterranean genetic isolate. Neurogenetics 16, 55–64 (2015). https://doi.org/10.1007/s10048-014-0425-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-014-0425-x