
Abstract
In this study, we retrospectively compared the prognostic value of the 2016 WHO classification with the former classification in 387 patients with glioma treated at our institution. According to the new classification, diagnoses included oligodendroglioma with isocitrate dehydrogenase (IDH) mutation and 1p/19q co-deletion (5.4%), anaplastic oligodendroglioma with IDH mutation and 1p/19q co-deletion (3.4%), diffuse astrocytoma IDH-mutated (3.9%), anaplastic astrocytoma IDH-mutated (2.8%), glioblastoma IDH-mutated (7.8%), glioblastoma IDH-wildtype (58.4%), diffuse midline glioma H3 K27M mutation (2.6%), oligodendroglioma NOS (1.3%), anaplastic oligodendroglioma NOS (0.8%), diffuse astrocytoma IDH-wildtype (2.8%), and anaplastic astrocytoma IDH-wildtype (10.9%). The prognoses of IDH-mutated astrocytomas clearly varied according to tumor grade. However, we identified no survival difference between IDH-wildtype anaplastic astrocytomas and glioblastomas; additionally, these tumors showed similar gene expression profiles. After exclusion of those without 1p/19q co-deletion, patients with oligodendroglial tumors showed excellent survival regardless of tumor grade. Our evaluation of chromosomal aberrations suggests that the MAPK/PI3K pathway plays a role in acquired malignancy of astrocytic tumors, whereas TP53 participates in tumorigenesis. We suspect the RB pathway also plays a role in tumorigenesis of IDH-mutated gliomas. The new WHO classification more clearly reflects the tumorigenesis of gliomas and improves the prognostic power of classification.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gliomas are the most frequent and malignant primary brain tumors. Diagnosis of gliomas has generally been based upon the phenotypes as determination by pathological examination specifically takes tumorigenesis into account.
The 2016 fourth update of the World Health Organization (WHO) classification of tumors of the central nervous system (CNS) [1] more clearly reflects tumorigenesis of glial tumors by classifying tumors based upon both their phenotypes and molecular features: isocitrate dehydrogenase (IDH) mutation, 1p/19q co-deletion, and H3 K27M mutation. With the incorporation of molecular diagnosis, this new classification decreases inter-observer discordance in classification, but has also created atypical categories of diagnosis: IDH-wildtype diffuse astrocytomas (DAs), IDH-wild-type anaplastic astrocytomas (AAs), oligodendrogliomas with no specific molecular features [oligodendrogliomas not otherwise specified (NOS)], and anaplastic oligodendrogliomas (anaplastic oligodendrogliomas NOS).
The aims of this study were to clarify the prognostic value of the 2016 WHO classification in clinical practice and to evaluate the nature of tumors classified into the atypical diagnostic categories according to their clinical and molecular characteristics.
Materials and method
Patients
All patients with gliomas treated in our institution were enrolled in this retrospective study of clinical data extracted from the patients’ medical records. The institutional review board approved the study protocol and waived the need for informed consent for our retrospective study, which was performed entirely at our institution.
Pathological diagnosis and immunohistochemistry (IHC)
Pathological diagnoses were made without molecular information by two pathologists (TS, MI) according to the 2007 WHO classification. In addition, automated immunohistochemistry (IHC) was performed on 4-µm-thick formalin-fixed, paraffin-embedded sections with an avidin–biotin–peroxidase complex on Dako Ominis (Agilent Technologies, Santa Clara, CA, USA) using a DAKO Kit including DAB reagent to search for expression of IDH1-R132H (clone H09) (Dianova GmbH, Hamburg, Germany).
DNA extraction, loss of heterozygosity (LOH) analysis, and mutation analyses of IDH1, IDH2 and H3F3A
Tumor DNA was extracted from frozen tissue, and normal DNA from peripheral blood leukocytes for all patients. Loss of heterozygosity (LOH) analysis was carried out at 12 loci on 1p and 4 loci on 19q: p73, D1S548, D1S450, D1S483, D1S2843, D1S458, D1S1676, D1S237, D1S2676, D1S2783, D1S496, MYCL1, D19S601, D29S867, D19S412, and D19S408. To analyze microsatellite alterations, specific primers with the 5′ labeled with FAM (tetrachloro-6-carboxyfluorescein) for the above markers were used in PCR of the amplified products. PCR products were loaded onto an ABI 3730 XL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) to determine the length distribution. Data were analyzed with Genemapper 4.0 (Applied Biosystems). The status of mutations of IDH1, IDH2, and H3F3A was addressed by direct sequencing using the Sanger method and primers, as described previously [2]. The methylation status of O-6-methylguanine-DNA methyltransferase (MGMT) gene promoter was evaluated by methylation-specific PCR analysis [3].
All tumors were also re-classified according to the 2016 WHO classification. In this study, tumors with oligodendroglial phenotype and 1p/19q co-deletion, but with intact IDH, were diagnosed as oligodendroglial tumors NOS.
Comparative genomic hybridization (CGH) arrays
Microarray-based comparative genomic hybridization (array-CGH) was performed with a DNA chip carrying 2464 BAC clones to examine genomic aberrations in 91 gliomas; chromosomal aberrations of the genes involved in the mitogen-activated protein kinase (MAPK)/phosphatidylinositol-3 kinase (PI3K) pathway (EGFR, PTEN, and NF1), p53 pathway (TP53), and RB pathway (CDKN2A and RB) were evaluated.
RNA extraction and c-DNA array
Tumor RNA was extracted from stored frozen tissue. Additionally, gene expression profiles of 42 gliomas were evaluated using 563 genes–cDNA–nylon microarrays [4], which were supplied by Kazusa DNA Research Institute (Kisarazu, Japan); these expression profiles were compared between the typical and atypical categories of diagnosis.
Statistical analysis
Overall survival after the first treatment was compared between categories of diagnosis. Kaplan–Meier analysis was used to draw survival curves and Cox proportional hazards analyses to examine factors associated with increased risk of death. Statistical analyses were performed using JMP for Mac V.11.0 (SAS Institute, Cary, NC, USA).
Results
Patients
From 1995 to 2017, 506 patients with gliomas were treated in our institution. Four patients with pilocytic astrocytomas were excluded from this study. Tumor samples for molecular analysis were not available in 115 cases; these patients were also excluded. Thus, 387 patients with gliomas were enrolled in this study. They comprised 149 female and 238 male patients, with a median age of 60 years (range 3–88 years). The median Karnofsky’s performance status at diagnosis was 70% (range 20–100%). The participants’ clinical characteristics are summarized in Tables 1.
IDH and H3F3A mutations
Expression of IDHR132H was investigated by IHC in 253 of 385 tumors, mutation status of IDH by direct sequencing in 269, and by both methods in 136. Expression of IDH1R132H protein was assessed in 51 of 253 tumors by IHC. IDH1 R132H, R132C, and IDH2 R172L mutations were identified in 34, 1, and 2 tumors, respectively, by direct sequencing. IDH mutations were not identified by IHC in one glioblastoma (GBM) with IDH1 R132H mutation, one DA with IDH1 R132C mutation, and two anaplastic oligodendrogliomas with IDH2 R172L mutation. There were no discrepancies between the results of IHC and direct sequencing in the other 132 tumors (97.1%).
H3 K27M mutations were found in 10 midline gliomas, including four GBMs, three AAs, two DAs, and one anaplastic oligodendroglioma. Their locations were pons in three, midbrain in two, cerebellum in one, and thalamus in four patients.
Loss of heterozygosity (LOH)
LOH of 1p was found in 133 tumors; however, deletion of all of the 12 loci on 1p was found in only 66 of them. LOH of 19q was found in 115 tumors; however, deletion of all the four loci on 19q was found in only 98 of them. Co-deletion of the whole arms of both 1p and 19q was identified in only 49 tumors.
Differences in diagnoses between the 2007 and 2016 WHO classifications
Reclassification of the tumors according to the 2016 WHO classification is summarized in Fig. 1. Only 21 of the 36 oligodendroglial tumors (58.3%) exhibited both an IDH mutation and 1p/19q co-deletion. The remaining 15 tumors were re-classified as oligodendroglioma NOS (five), IDH-mutated DA (five), and IDH-wildtype DA (five). Similarly, only 13 of the 44 anaplastic oligodendroglial tumors (29.5%) exhibited both an IDH mutation and 1p/19q co-deletion. The remaining 31 tumors were re-classified as anaplastic oligodendroglioma NOS (three), IDH-mutated AA (five), IDH-wildtype AA (22), and diffuse midline glioma with H3 K27M mutation (one).

Reclassification of all tumors according to the 2016 WHO classification
Mutations of IDH were found in only 16 of the 47 lower grade astrocytomas. H3 K27M mutations were identified in five of them; the remaining 26 tumors were classified into the atypical categories of diagnosis, IDH-wildtype DAs, and AAs. H3 K27M and IDH mutations were found in 4 and 30 of the 260 glioblastomas, respectively.
Prognostic value of former and new WHO classifications
During a median follow-up of 2.4 years (range 0.0–20.9 years), 279 of the patients had died. The median survival time of all patients was 1.7 years (95% CI 1.5–1.9 years) after the first treatment.
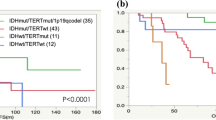
Using the former classification of astrocytic tumors, survival of patients with AA (median 1.3 years, 95% CI 1.1–1.5) did not clearly differ from that of those with GBM (median 1.3 years, 95% CI 1.1–1.5) (HR 1.53, 95% CI 0.98–2.54, P = 0.060); however, survival of patients with DA (median not reached, 95% CI 4.4 years-not reached) was significantly superior to that of these patients (Fig. 2A). In contrast, using the 2016 WHO classification, Kaplan–Meier survival curves showed clear prognostic differences according to tumor grade (II, III, and IV) between astrocytic tumors with IDH mutations (Fig. 2C). In this molecular subsets of tumors, survival of patients with grade III (median 4.1 years, 95% CI 2.2–10.3) and IV tumors (median 1.2 years, 95% CI 0.9–2.2) differed significantly (HR 2.98, 95% CI 1.55–6.58, P = 0.005); however, likely because of the small number of patients, there was no significant difference in survival between grade II (median: not reached, 95% CI 4.4-not reached) and III tumors (HR 2.64, 95% CI 0.83–9.92, P = 0.099). In our series, the prognostic value of IDH mutations was not proven for grade IV tumors (HR 0.99, 95% CI 0.66–1.58, P = 0.994). Patients with atypical categories of tumors according to the new classification showed poorer survival after treatment. Survival of patients with IDH-wildtype AA (median 1.6 years, 95% CI 0.9–2.4) was significantly poorer than that of those with IDH-mutated AA (HR 2.40, 95% CI 1.17–5.60, P = 0.016), and similar to that of those with GBM (Fig. 2C). Survival of patients with IDH-wildtype DA (median 24.3 years, 95% CI 1.3–24.3) was also poorer than that of those with tumors with IDH mutations and similar to that of those with grade III tumors with IDH mutations, the latter difference not being significant likely being attributable to the small number of patients (HR 1.75, 95% CI 0.46–7.08, P = 0.402, Fig. 2C, Table 2).

Overall survival curves according to the former classification (A) and the updated 2016 WHO classification (B–D). Survivals of typical categories of tumors (B), astrocytic tumors (C), and oligodendrogliomas (D) according to the new classification have been drawn separately. AA anaplastic astrocytoma, AO anaplastic oligodendroglioma, AOA anaplastic oligoastrocytoma, DA diffuse astrocytoma, GBM glioblastoma multiforme, IDHm IDH-mutated, IDHw IDH-wildtype, O oligodendroglioma, OA oligoastrocytoma
Using the new WHO classification, the prognostic value of the category “oligodendroglial tumors” became more significant because it eliminates tumors with both IDH mutation and 1p/19q co-deletion (Fig. 2A, B). The median survival times of patients with oligodendroglial and astrocytic tumors were 6.2 and 1.5 years, respectively (HR 3.0, 95% CI 2.2–4.2, P < 0.0001), in the former classification, and 17.0 and 1.5 years, respectively (HR 5.3, 95% CI 3.2–9.5, P < 0.0001), in the new classification. However, we did not identify any survival difference between patients with grade II (median 17.0 years, 95% CI 10.8–not reached) and III tumors (median: not reached, 95% CI 2.1–not reached) (HR 1.06, 95% CI 0.23–3.83, P = 0.485). The number of oligodendroglial tumors NOS was too small to analyze statistically; however, oligodendroglial tumors with inconclusive molecular findings showed poorer survival after diagnosis (Fig. 2D, Table 2).
MGMT promoter methylation
Methylation status of MGMT gene promoter was available for 348 tumors. The majority (90.5%) of oligodendroglial tumors with both the IDH mutation and 1p/19q co-deletion exhibited methylation, whereas only half the tumors NOS showed methylation. No difference in frequency of methylation according to grade of tumor was found for astrocytic tumors, whereas those with IDH mutilations more frequently demonstrated methylation (60.0%) than IDH-wildtype tumors (38.8%, P = 0.009, Table 1).
Copy numbers of EGFR, PETN, NF1, TP53, CDKN2A, and RB1 genes
Frequencies of alterations in copy number of EGFR, PETN, NF1, TP53, CDKN2A, and RB of 86 gliomas are summarized in Fig. 3.

Frequencies of copy number alterations of EGFR, PTEN, NF1, TP53, CDKN2A, and RB. Mut mutation
Increased or decreased copy numbers of any one of the three genes involved in the MAPK/PI3K pathway were found in 83.7% of IDH-wildtype GBMs, 75.0% of IDH-mutated GBMs, 33.3% of IDH-mutated AAs, and 25.0% of IDH-mutated DAs. Alterations in copy numbers of these genes in astrocytic tumors were more frequently observed in grade IV than lower grade tumors with IDH mutations (P = 0.014), whereas IDH-wildtype AAs showed a high frequency of copy number changes (70.0%). Regardless of grade, 40.0% of oligodendroglial tumors with both 1p/19q co-deletion and IDH mutation showed these changes, whereas 60.0% of oligodendroglioma NOS exhibited these alterations.
Decreased copy numbers of TP53 were not observed in grade II astrocytic tumors, but were found in grade III and IV tumors. There was no difference in frequency of decreased copy numbers of this gene between GBMs and AAs, these tendencies being observed regardless of the status of IDH mutation. Alteration of this gene was more frequent in oligodendroglial tumors, also being observed in grade II tumors. There was no difference in frequency of alteration of this gene between typical oligodendroglioma and oligodendroglioma NOS.
Decreased copy numbers of genes involved in the RB pathway were found in 46.9% of IDH-wild type GBM and were more frequently observed in IDH-mutated astrocytic tumors regardless of grade: 100.0% in grade IV, 66.7% in grade III and 75.0% in grade II tumors (P = 0.048). The frequency of alterations in lower grade tumors without IDH mutation (36.3%) was similar to that of IDH-wildtype GBMs. Decreased copy numbers of these genes were found in 60.0% of typical oligodendroglial tumors regardless of grade. Oligodendroglioma NOS showed more frequent alteration of these genes than typical oligodendrogliomas; however, this difference was not significant.
Gene expression profile
The strength of expression of 221 genes was found to differ significantly between oligodendroglial and astrocytic tumors. The expression profiles of these 221 genes were evaluated in 42 gliomas by clustering analysis (Fig. 4). The 221 genes included five proneural and four mesenchymal signature genes derived by Phillips et al. [5]. The expression profiles of typical oligodendrogliomas and IDH-wildtype GBMs were quite different and all of the proneural genes were included in the genes strongly expressed in typical oligodendrogliomas and mesenchymal genes strongly expressed in IDH-wildtype GBMs. The expression profiles of oligodendroglioma NOS were similar to those of typical oligodendrogliomas, whereas the expression profiles of IDH-mutated AAs and GBMs were similar to those of oligodendrogliomas rather than IDH-wildtype GBMs and the expression profiles of IDH-wildtype AAs were similar to those of IDH-wildtype GBMs.

Clustering of 42 gliomas according to expression of 221 genes
Discussion
In this study, we re-classified 387 gliomas according to the 2016 WHO classification of brain tumors. The distribution of subgroups of gliomas in this single institutional study differs from the reported data for gliomas in Japan [6]: the percentages of DA and AA were lower and that of GBM higher in our series.
Deletions of chromosomes 1p and 19q are reportedly closely associated with oligodendroglial tumors [7, 8]. We have previously reported that long terminal deletion of 1p, not interstitial small deletion, is a prognostic marker in oligodendroglial tumors [9]. More recently, the mechanism for concurrent loss of 1p and 19q had been shown to be translocation of chromosomes 1 and 19 at the centromere [10, 11]. Deletions of the whole arms of 1p and 19q are required to diagnose oligodendrogliomas. In clinical practice, fluorescence in situ hybridization (FISH) is the preferred means of diagnosing 1p/19q co-deletion; however, only one locus on each arm is checked by this method. In our series, we found LOH of 1p in 37.3% of the investigated tumors and whole deletion in only half of them (49.6%), indicating the limitations of using FISH to diagnose 1p/19q co-deletion.
Genetic driver mutations in the IDH1 and IDH2 are closely associated with gliomagenesis [12]. Among the three major mutations of IDH: IDH1 R132H, IDH1 R132C, and IDH2 R172L, IDH1 R132H occur most frequently; 67 of 70 IDH-mutated tumors (95.7%) showed this type of mutation, whereas we identified only one IDH1 R132C and two IDH2 R172L mutations. IHC of IDH1R132H proteins is sufficient to assess the mutation status of IDH in clinical practice. Among the 136 tumors for which both IHC and direct sequencing were available, we found concordance between these two variables in 99.2% (132/133) of tumors with IDH1 R132H mutations; this frequency is similar to that reported previously [13, 14]. IHC cannot diagnose minor mutations of IDH; however, the sensitivity and specificity of IHC was 83.3% and 100.0%, respectively, for diagnosing IDH mutations in our cases.
In our study, numerous oligodendroglial tumors did not exhibit 1p/19q co-deletion; accordingly, their diagnoses were switched to astrocytic tumors to conform to the new classification. This phenomenon occurred significantly more frequently with mixed gliomas (P = 0.036) and grade III tumors (P = 0.001). The prognoses of patients with these tumors were poorer than those of patients with typical oligodendroglial tumors and the prognostic power of classification (oligodendroglial versus astrocytic tumors) was greater with the new classification.
We found that prognoses of patients with IDH-mutated astrocytomas to be clearly associated with grade. In this type of tumor, the frequency of alterations in copy numbers of the genes involved in the MAPK/PI3K pathway increased with grade, indicating that these genes are associated with acquired malignancy of tumors in this lineage. EGFR amplification is reportedly specific for primary GBMs, whereas PTEN loss occurs in both primary and secondary GBMs [15]. In IDH-wildtype GBMs, changes in copy numbers of these genes were more frequent (83.7%); however, in our series IDH-mutated GBMs also frequently exhibited changes in these genes (75.0%). This frequent genomic change may in part explain why patients in our series with IDH-mutated and IDH-wildtype GBMs showed similar survival after diagnosis. In contrast to EGFR, we found no difference in frequency of TP53 loss between grade III and IV astrocytomas, suggesting that alteration of this gene is an early event in the tumorigenesis of these tumors. Genomic alterations of the RB gene are reportedly frequently present in secondary GBMs [16]. In our series, we more frequently observed decreased copy numbers of genes involved in the RB pathway in IDH-mutated astrocytic tumors and IDH-mutated oligodendroglial tumors than in IDH-wildtype astrocytic tumors, indicating that the RB pathway may participate in tumorigenesis of IDH-mutated tumors.
In the 2016 WHO classification, secondary GBM lineage is classified into IDH-mutated astrocytomas and primary GBM into IDH-wildtype GMB on the basis of gliomagenesis, whereas IDH-wildtype DA and AA are not classified into a known lineage of tumors. Patients with IDH-wildtype DA have similar survival to those with IDH-mutated AA, and those with IDH-wildtype AA have similar survivals to those with GBMs. Our genomic investigation revealed that the frequency of changes in copy numbers of genes involved in the MAPK/KI3K pathway in IDH-wildtype AA (70.0%) is similar to that in IDH-mutated and -wildtype GBMs (83.7% and 75.0%, respectively) rather than in IDH-mutated AA (33.3%). Furthermore, we found that the expression profiles of IDH-wildtype AA are similar to those of IDH-wild type GBMs. These clinical and molecular features indicate the more aggressive character of these tumors and their requirement for intensive treatment.
Patients with oligodendroglial tumors with both the 1p/19q co-deletion and IDH mutation demonstrated excellent survival after diagnosis. The five-year survival rate of patients with grade III tumors was 83.2% (95% CI 55.9–95.1) in our series, which is similar to previously reported French data [12]. Long-term risk and benefit of treatments for oligodendroglial tumors diagnosed according to the new classification should be precisely elucidated in the light of these excellent survivals. We were unable to draw any conclusions about oligodendroglial tumors NOS because there were so few patients with them in our series. However, these tumors exhibited more frequent alterations in copy numbers of genes involved in the MAPK/PI3K and RB pathways and the gene expression profiles of some of them were more similar to those of GBMs than those of typical oligodendrogliomas, indicating that tumors in this category of gliomas may be still heterogeneous and affected patients, therefore, require careful management during and after treatment.
The major limitation of this study was the biased distribution of the tumors, which included many IDH-wildtype GBMs, but only a few tumors in the other categories. Despite this bias of distribution, we found that the 2016 WHO classification was of greater prognostic value than the former classification and would, therefore, better enable optimal treatment choices for individual patients in clinical practice. Further investigation, especially of oligodendroglioma NOS, is required.
References
Louis DN, Ohgaki H, Wiestler OD et al (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109
Louis DN, Perry A, Reifenberger G et al (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131:803–820
Hegi ME, Diserens AC, Gorlia T et al (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352:997–1003
Yamakawa H, Yokoyama S, Hirano T et al (2004) A simple and robust method for preparation of cDNA nylon microarrays. DNA Res 11:353–360
Phillips HS, Kharbanda S, Chen R et al (2006) Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9:157–173
(2017) Brain tumor registry of Japan (2005–2008). Neurol Med Chir (Tokyo) 57(Supplement-1): 9–102
Ransom DT, Ritland SR, Kimmel DW et al (1992) Cytogenetic and loss of heterozygosity studies in ependymomas, pilocytic astrocytomas, and oligodendrogliomas. Genes Chromosomes Cancer 5:348–356
Bello MJ, Vaquero J, de Campos JM et al (1994) Molecular analysis of chromosome 1 abnormalities in human gliomas reveals frequent loss of 1p in oligodendroglial tumors. Int J Cancer 57:172–175
Iuchi T, Namba H, Iwadate Y et al (2002) Identification of the small interstitial deletion at chromosome band 1p34-p35 and its association with poor outcome in oligodendroglial tumors. Genes Chromosomes Cancer 35:170–175
Jenkins RB, Blair H, Ballman KV et al (2006) A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res 66:9852–9861
Griffin CA, Burger P, Morsberger L et al (2006) Identification of der(1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J Neuropathol Exp Neurol 65:988–994
Tabouret E, Nguyen AT, Dehais C et al (2016) Prognostic impact of the 2016 WHO classification of diffuse gliomas in the French POLA cohort. Acta Neuropathol 132:625–634
Yan H, Parsons DW, Jin G et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773
Sanson M, Marie Y, Paris S et al (2009) Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 27:4150–4154
Ohgaki H, Dessen P, Jourde B et al (2004) Genetic pathways to glioblastoma: a population-based study. Cancer Res 64:6892–6899
Gladson CL, Prayson RA, Liu WM (2010) The pathobiology of glioma tumors. Annu Rev Pathol 5:33–50
Acknowledgements
We thank Dr Trish Reynolds, MBBS, FRACP, from Edanz Group (http://www.edanzediting.com/ac) for editing a draft of this manuscript. This study was partially supported by a JSPS KAKENHI Grant (Grant No. 15K10349).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Iuchi, T., Sugiyama, T., Ohira, M. et al. Clinical significance of the 2016 WHO classification in Japanese patients with gliomas. Brain Tumor Pathol 35, 71–80 (2018). https://doi.org/10.1007/s10014-018-0309-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10014-018-0309-0