
Abstract
Acid–base equilibria are generally studied and taught at universities using approaches and techniques that include the use of dyes, spectrophotometry, conductometry, and potentiometry. Instead, voltammetric techniques, although employed for research purposes for acid–base investigations, have rarely been included in electrochemical curricula. In this article, we highlight the potential of microelectrode voltammetry in studying acid–base equilibria, their kinetics, and the acid and base content in aqueous solutions by exploiting the hydrogen and oxygen evolution reactions. Microelectrodes are used as they allow the attainment of reproducible and well-defined convergent mass-transport conditions and the achievement of steady-state diffusion regimes in short times. The resulting steady-state limiting current is proportional to bulk concentration, diffusion coefficient, and electrode radius, which is useful for a more precise evaluation of each of latter quantities. Mention is also made on how mathematical treatments and digital simulation procedures can help in the classification and parameterization of the electrode processes involved.
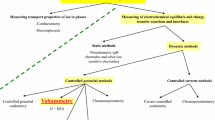
Graphical abstract
Hydrogen and oxygen evolution from neutral and acid or base aqueous solutions.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
An integral part of chemistry courses taught at universities and colleges is the study of acids and bases and their equilibria [1, 2]. Acids and bases [3] are important classes of chemical substances, as they are involved in physiological functions [4], in food stability, and in many areas of industrial chemistry and synthesis of a variety of compounds [5,6,7]. Although acids and bases had intrigued observers of nature since antiquity, their understanding has been shaped by the progress of chemistry [1, 2, 4, 8]. Originally, acids and bases were identified by the taste of their aqueous solutions (i.e., sour and bitter, respectively) and the ability to change colors to solutions containing certain substances, as for example the natural dye litmus (from blue to red for acids and red to blue for bases) [2, 8]. The conceptual evolution of understanding acid–base characteristics is inseparably intertwined with the history of chemistry. Prodromal to the theory for acids and bases were the studies regarding salts, electrolyte dissociation in water solutions, cations and anions, and the observation that passing electricity through water it was split into its constituent elements [9]. It was in this context that Nobel laureate Svante Arrhenius defined an acid as a compound that when dissolved in water yields hydrogen cations and a base as one that when dissolved in water yields hydroxide anion [10, 11]. Meanwhile, the Danish chemist Søren Peter Lauritz Sørenson introduced in 1909 the concept of pH (the negative power of 10 for the concentration of hydrogen ion) as a scale for measuring acidity and alkalinity [11]. A more general theory of acids and bases was proposed by two physical chemists, the Danish Johannes N. Brønsted and the English Thomas M. Lowry, who extended the definition into solvents other than water by characterizing acids as proton donors and bases as proton acceptors independent of their solvent solution [1, 2, 4, 8, 9, 12, 13]. In the same period, the American physical chemist, Gilbert N. Lewis, presented a broader definition of acids and bases, also independent of their solvent solution, by characterizing acids as an electron-deficient, i.e., acceptor species, and base as an electron donor species [4, 14, 15]. Among these models, the Brønsted–Lowry (B-L) concept represents a practical and useful compromise between the classical but too restrictive Arrhenius concept and the somewhat too general Lewis concept, especially from an analytical point of view.
Considering B-L theory, acidity (or basicity) of a medium and the acid–base equilibria are generally studied and taught using approaches and techniques that include the use of dyes (i.e., in classical volumetric titrations), spectrophotometry and, among electrochemical techniques, conductometry and potentiometry. Several old [1, 16, 17] and more recent [2] books deal with these aspects, where fundamentals are illustrated for an understanding of acid–base equilibria involved in acid–base titrations, on how to obtain parameters such as pH (or pOH), acidity or basicity constants, and total acid content in a medium. Laboratory experiments for undergraduate students, aimed at supplementing current textbook examples, have also proposed in lecture-type articles [18,19,20].
Earlier before 1960s, studies on acid–base equilibria have also been performed by using polarography (i.e., the use of mercury as the working electrode) [21, 22] and voltammetry at rotating disk electrodes [23, 24] to mainly obtain information on thermodynamic and kinetic constants. In the early eighties and later, with the advent of electrodes having micrometer dimensions or less (i.e., microelectrodes, also commonly known as ultramicroelectrodes) [22, 25,26,27,28,29], voltammetric studies on acid–base systems have also been extended to obtain information on the acid and base content in a variety of solutions. Nowadays, voltammetry and microelectrodes have become topics in electrochemical and electroanalytical teaching curricula. However, they mainly deal with the general aspects of the techniques and with some applications for the quantitative determination of analytes [30, 31]. Instead, the possibility offered by these techniques, as alternative to potentiometry and conductometry, for studying acid–base base equilibria or probing the acidity (or basicity) levels of a medium, is almost ignored. Therefore, herein, we highlight how information on the acid–base properties of aqueous solutions can be obtained by microelectrode voltammetry, exploiting the hydrogen and oxygen evolution reactions. Mention is also made to the mechanisms involved in the electrode processes and how mathematical treatments and digital simulation procedures can help in their classification and parametrization. Microelectrodes are considered as they allow the attainment of reproducible and well-defined convergent mass-transport conditions and the achievement of steady-state diffusion regimes in short times [22, 25,26,27,28,29].
The examples presented in the article can be useful for teachers who want to implement electrochemistry-based laboratory experiments to improve the students’ ability to understand the existing relationships between currents, due to interfacial electron transfer, mass transfer, and chemical reactions in the homogeneous phase coupled with the electron transfer process. In addition, it is also highlighted how mathematical and simulation approaches can help in the deeper understanding of the phenomena involved.
Acid–base equilibria and parameters based on the B-L concept
According to the (B-L) model, a generic acid–base equilibrium can be written as given by equation T1 (Table 1). For a generic acid HA or base B in water solutions, relevant protolysis equilibria can be expressed by reactions T2 and T3, respectively (Table 1), where \(\mathrm A^-\) is the base conjugate to the acid HA and \(\text{BH}^+\)is the acid conjugate to the base B. According to reactions T2 and T3, H2O can act either as a base or an acid. The strength of the acid or base, measured by the acidity or basicity constants, Ka and Kb, respectively, is expressed by the relationships shown in Table 1. In water, Ka and Kb are related through the water autoprotolysis constant Kw, given by equation T4. It must be noted that the relationships of thermodynamic constants should be expressed in terms of activity. Here, the constants due to the protolysis of HA and B are provided in terms of concentrations, as it is assumed that their values are evaluated at a constant ionic strength (different from 0) [1, 2].
The study of the acid–base equilibria typically involves the knowledge of the acidity and basicity constants, the kinetic involved in the dissociation and recombination reactions, concentrations of all species at equilibrium, pH or pOH, as well as the analytical (i.e., total) concentrations of the acid (CA) or base (CB).
Voltammetry under planar and radial diffusion
Voltammetry is a term used to indicate a variety of electroanalytical techniques in which the current is measured as a function of voltage [30, 31]. Among voltammetric techniques, linear sweep voltammetry (LSV) and cyclic voltammetry (CV) are the most popular techniques employed by electrochemists for general investigations [22, 30, 31, 33]. In this article, we consider LSV, characterized by the waveform shown in Fig. 1a. The current against potential response depends on whether planar (Fig. 1b) or radial (Fig. 1c) diffusion occurs. The different conditions can be obtained, respectively, by using so-called conventional electrodes (or macroelectrodes) [22, 25,26,27,28,29, 33], i.e., electrodes having, typically, size in the millimeter dimensions [22] and microelectrodes, characterized by at least one dimension (e.g., radius of a disk or a fiber and width of a band, called critical dimension) smaller than 25 µm [22, 25,26,27,28,29, 33]. Figure 1 includes typical voltammograms recorded for a generic reversible reduction process Ox + ne = Red, at a millimeter disk (Fig. 1d) and a micrometer disk (Fig. 1e) electrode. The peak current (Ip in amperes) of the LSV response at conventional electrodes at 25 °C is given by [22]
where F is Faraday constant (exactly, 9.64853321233100184 × 104 C⋅mol−1) [34], n is the number of exchanged electrons, A is the electrode surface area (cm2), D (cm2 s−1) and C (mole cm−3) are the diffusion coefficient and the bulk concentration of the electroactive species, respectively, and v (V s−1) is the scan rate. The steady-state diffusion limiting current (Il in Fig. 1) at a microdisk electrode is given by [35]
where a is the radius of the microdisk, other symbols have the meaning as above. It must be noted that at microelectrodes, radial diffusion applies when the thickness of the diffusion layer is larger compared to the critical dimension of the electrode [25]. This occurs at relatively long times, at constant applied potentials, or at low scan rates in potential sweep methods [22, 36]. Under such conditions, a steady-state current is achieved in short times. Comparing Eqs. (1) and (2), an important consideration can be done on the dependence of D on the experimentally measured quantities (e.g., current or electrode dimension). Under conditions of linear diffusion, Ip is proportional to D1/2 and A (i.e., π r2), whereas under steady-state condition at the disk microelectrode Il is proportional to D and a. Therefore, D depends on Ip2 and Il using macro- or microdisk electrodes, respectively. This implies that any errors on measured current produce a smaller error on the evaluation of the diffusion coefficient using microelectrodes [27].

LSV waveform (a); Ei and Efin are the initial and final potential, respectively. Diffusion field at a macroelectrode (b) and microelectrode (c). Voltammetric responses and typical parameters at a macroelectrode (d) and at a microelectrode at low scan rates (e)
The use of voltammetric methods to investigate electrode reaction mechanisms and their kinetics typically requires the analysis of experimental responses measured under only diffusion conditions as a function of the potential scan rate, in the case of macroelectrodes, or as a function of the electrode size in the case of the steady-state measurement at microelectrodes [29, 33].
Voltammetry in aqueous media at noble metal electrodes
In neutral deaerated aqueous solutions, e.g., those containing supporting electrolytes such as NaClO4, LiClO4, KNO3, and Na2SO4, the negative and positive potential limits at noble metal electrodes, such as platinum and gold, are due to the reduction and oxidation of water, involving the hydrogen (reaction 3) and oxygen (reaction 4) evolution process, respectively:
where \(\mathrm H^+\) (aq) represents the hydrated hydronium \((\text{H}_3\text{O}^+)\). Typical LSV responses at Pt and Au microelectrodes are shown in Fig. 2. Other electrolyte, such as KCl (often employed as supporting electrolyte), in the positive potential region, can give rise to the oxidation process of the anion, which can occur before or concomitantly to reaction (4).

LSVs recorded in an aqueous solution containing LiClO4 0.1 M, at Pt (red line) and Au (black line) disk microelectrodes, 12.5-µm radius, scan rate 5 mV s−1
As will appear clear in what follows, in dilute aqueous solutions of acids and bases, the hydrogen and oxygen evolution reactions provide voltammetric responses, which are separated from the background discharge. From the current intensity, the potential at which the process takes place and the shape of the voltammetric wave, information on the acid or base strength, kinetics involved in the equilibria, and the acid or base concentrations can be obtained. Examples are illustrated for some most common strong and weak acids and bases. Most of the work presented here relies on responses obtained by using microdisk electrodes and LSV at low scan rates.
Hydrogen evolution from strong acid solutions at platinum electrodes
The simplest case of a generic monoprotic strong acid is considered first, as in aqueous solution it is fully dissociated HA + H2O → H3O+ + A−, and the hydronium reduction process can be written as
At Pt electrodes, despite the complexity of the mechanism, which involves adsorption and recombination steps, the reduction process of H3O+ can be assumed to be reversible [37,38,39,40]. However, to obtain reproducible responses, mechanical and electrochemical cleaning of the electrode surfaces is needed [37,38,39,40,41,42,43]. The mechanical treatment consists in polishing the electrodes with successively finer grades of alumina (typically, from 5 µm down to 0.3 µm) suspended in water on suitable microcloths. The electrochemical pretreatment consists in cycling the potential several times over potential regions covering the Pt oxide/hydroxide formation and reduction [37,38,39,40].
Using Pt microelectrodes, having radii around 12.5 µm (or less), steady-state voltammograms as those shown in Fig. 3a are obtained. They refer to LSVs recorded at a 12.5 µm Pt disk electrode at 5 mV s−1 in aqueous solutions containing HClO4 at different concentrations and 0.1 M KCl as supporting electrolyte. All strong acids (e.g., HCl and HNO3) at the same CA provide overlapping waves, as only H3O+ feeds the electrode process. The well-shaped sigmoidal waves in Fig. 3a reflect the prevailing of radial diffusion and the achievement of steady-state conditions. Therefore, the plateau current is given by
where \({D}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}\) and \({C}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}\) are the diffusion coefficient and bulk concentration of \(\text{H}_3\text{O}^+\), the number of electrons n being equal to 1, other symbols have their usual meaning. Equation (6) predicts a linear dependence between Il and \({C}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}\) and consequently also with CA (Fig. 3b). The calibration plot can be used to detect an unknown concentration of \(\text{H}_3\text{O}^+\) of a strong acid. It can also be exploited to obtain \({D}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}\) provided that all other parameters of Eq. (6) are known. From the slope of the plot shown in Fig. 3b, a value of 7.92 × 10−5 cm2 s−1 [37] was evaluated, which is within other quoted values reported in the literature at the stated temperature of 25 °C (Table 2). Differences in \({D}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}\)can be due to the specific experimental conditions employed in the measurements, for instance, on the actual temperature, on the nature and concentration of the supporting electrolyte, which affect the viscosity and ionic strength of the medium [44]. These parameters affect the diffusion coefficient according to the Stokes–Einstein law [45]:
where k is the Boltzmann constant, T is the temperature (in K), and rh is the hydrodynamic radius. For diffusion coefficient measurements, the radius of the microdisk needs to be well calibrated, and this can be done by scanning electron microscopy (SEM) (see image in inset Fig. 3a), or by steady-state voltammetry using redox couples of known electrochemistry (e.g., Ru(NH3)63+/2+ [46]), together with Eq. (6).

a LSV responses recorded in 0.1 M KCl aqueous solutions containing 0.3 mM (red line) and 0.5 mM (blue line) HClO4, with a 12.5 µm radius Pt microdisk, scan rate 5 mV s−1 (adapted from Ref [47]. with permission); inset in a SEM image of the surface of a Pt microdisk; b Il vs. CA, c log Il vs. pH, and d \({E}_{1/2}\) vs. log CA plots obtained in 0.1 M KCl aqueous solutions containing HClO4 at different concentrations
The acid concentration is related to pH of the solution through equations T5–T6, and considering that Eq. (6) relates the limiting current to the hydronium concentration, a relationship between pH and Il or vice versa can be derived:
A linear dependence exists between \(\mathrm{log}\;{ I}_{\mathrm{l}}\) and pH with a slope equal to −1. This is shown in Fig. 3c for the simultaneous measurements of Il and pH, performed in a series of HClO4 solutions at different concentrations in 0.1 M KCl as supporting electrolyte. Provided that all other parameters are known, Eq. (7) can therefore be employed to obtain pH from steady-state limiting currents or vice versa.
Voltammetric reversibility of the H3O+/H2 couple
From the entire voltammogram, the potential parameters half-wave potential (\({E}_{1/2})\), i.e., the potential at half of the steady-state limiting current, inset in Fig. 1e, and Tomeš difference \(({E}_{1/4}-{E}_{3/4})\) [55] (i.e., the difference between the potentials at ¼ and ¾ of the steady-state limiting current, see Fig. 1e) can be obtained. \({E}_{1/2}\) is related to the equilibrium potential of the redox couple and \(({E}_{1/4}-{E}_{3/4})\) provides information on the reversibility of the electrode process [56].
A specific characteristic of reaction (5) is the nonunity stoichiometry, specifically 2:1, which is worth to be considered, as it influences the voltammetric responses. Firstly, unlike the electrode reactions of 1:1 stoichiometry (e.g., like that of the Ru(NH3)63+/2+ redox couple [46]), in which \({E}_{1/2}\) is unaffected by the change of the bulk concentration of the reactant specie, for reaction (5), \({E}_{1/2}\) depends on \(\mathrm{ln}{C}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}\) according to the following relationship [37,38,39, 57, 58]:
where \({E}^{0,\mathrm{c}}\) is the reference potential of the H3O+/H2 system for unity activity of both forms of the redox couple in the solution given by [38, 39]
\({E}_{{\mathrm{H}}_{3}{\mathrm{O}}^{+}/{\mathrm{H}}_{2}}^{0}\) is the normal hydrogen electrode potential (NHE), \({D}_{{\mathrm{H}}_{2}}\) is the diffusion coefficient of H2, \({C}_{{\mathrm{H}}_{2}}\) is the hydrogen concentration corresponding to the hydrogen partial pressure, \({P}_{{\mathrm{H}}_{2}}\), at 1 bar, and the other symbols have their usual meaning. Equation (8) indicates that \({E}_{1/2}\) shifts toward less negative potentials by 29.6 mV for a decade decimal logarithm variation (ddl) of \({C}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}\) at 25 °C. Experimental data points obtained from LSVs recorded in the series of measurements recorded in HClO4 solutions at different concentrations agree with this prediction (Fig. 3d). Secondly, the nonunity stoichiometry of reaction (6) also affects the steepness of the steady-state voltammogram, which can be accounted for the \(({E}_{1/4}-{E}_{3/4})\) parameter. In this case \(({E}_{1/4}-{E}_{3/4})\)= 42 mV [37,38,39], which differs from 56.4 mV expected for a reversible electrode process with a 1:1 stoichiometry [56].
Hydrogen evolution process from weak acids
For a monoprotic weak acid, the hydrogen evolution process can occur through the so-called CE mechanism (i.e., chemical reaction in the homogeneous solution that precedes the heterogeneous electron transfer process) [22]:
In such cases, the voltammetric responses is affected by the nonunity stoichiometry of the hydrogen evolution reaction, as well as by the kinetic of the acid dissociation, that is, on the first-order forward, k1, and second-order backward, k2, rate constants. These are related to the acidity constant through Ka = k1/k2. Since the recombination (backward) rate constant is usually diffusion-controlled (i.e., k2 ~ 1 × 1010 M−1 s−1 [59]), k2 has about the same value for various weak acids (Table 3). Therefore, the kinetic of the acid dissociation reaction depends, essentially, on k1 (Table 3) and ultimately on Ka.
Due to the high values of k1 and k2, protolytic reactions in aqueous solutions of most weak acids belong to the class of very fast processes [2, 59], and reaction (10) is under a mobile equilibrium. This behavior is common for monoprotic weak acids with Ka/CA > 10−3 and when the microelectrodes have radii ≥ 5 μm [37, 38, 43, 52, 60, 61]. When Ka/CA < 10−3 or the microdisks are very small, the kinetics of the dissociation reaction to form H3O+ becomes a significant factor. Furthermore, when the acid dissociation is too slow, the undissociated HA can directly be reduced to hydrogen through the following alternative path [43, 52, 60, 62]:
It must be considered that the latter mechanism rarely occurs in aqueous media at Pt electrodes as the water reduction hides all other processes (see below). Examples on how the different situations can be identified are described below.
Typical steady-state voltammograms recorded at a Pt electrode 12.5-µm radius, for a series of weak acids, and for comparison that of a strong acid, at the same 1 mM analytical concentration, in aqueous solutions containing 0.1 M KCl as supporting electrolyte, are shown in Fig. 4a. The acid strength affects the voltammetric parameters Il, \({E}_{1/2}\), and \(({E}_{1/4}-{E}_{3/4})\). In particular, at a given CA, Il decreases, \({E}_{1/2}\) shifts toward more negative potentials (Fig. 4b, black symbols), and \(({E}_{1/4}-{E}_{3/4})\) increases (Fig. 4b, blue symbols) as Ka decreases. These facts imply that the reduction waves of weak acids of low Ka are shifted to more negative potentials and flatten to such an extent that their processes cannot be distinguished from the background current or the water discharge process. This is the case, for instance, of HA with pKa ~ 9 or higher [37, 60, 62, 69]. The effect of pKa on the \(({E}_{1/4}-{E}_{3/4})\) parameter, which increases by increasing pKa, is explained considering that both H3O+ and HA diffuse toward the electrode surface, and therefore, their different diffusivities, concentrations at equilibrium, and acid dissociation kinetics play a role in the overall reduction process.

a LSV responses recorded at a Pt microelectrode, 12.5-µm radius, in 0.1 M KCl aqueous solutions containing 1 mM of the weak acids: \({\mathrm H}_2{\mathrm{PO}}_{4^-}\) (pink line); CH3COOH (blue line); ClCH2COOH (green line); HClO4 (red line); background discharge from 0.1 M KCl solution (black dotted line); scan rate 5 mVs−1 (adapted from Ref. [37] with permission). b \({E}_{1/2}\) vs. pKa (black symbols) and \(({E}_{1/4}-{E}_{3/4})\) vs. pKa (blue symbols) from a range of weak acids. c Il vs. CA plots for the weak acids: \({\mathrm H}_2{\mathrm{PO}}_{4^-}\) (red line and symbols); CH3COOH (blue line and symbols); lactic acid (black line and symbols) (adapted from Ref. [37] with permission); the full lines correspond to theoretical plots obtained by Eq. (15). d \({E}_{1/2}\) vs. log CA for a range of acids with different pKa, as indicated in the figure
The effect of CA on the wave height and position is of interest from both an analytical and kinetic point of view. Figure 4 includes plots of Il vs. CA (Fig. 4c, symbols) and \({E}_{1/2}\) vs. log CA (Fig. 4d, symbols) for a range of weak acids. As is evident, for the weak acids, Il is not strictly linear over a wide concentration range, while \({E}_{1/2}\) shifts toward either less negative or more negative potentials by increasing CA, depending on pKa of the acid. In particular, \({E}_{1/2}\) shifts toward less negative potentials for the acids with pKa < 3.6 (i.e., hydrogen sulfate ion and chloroacetic acid), similarly to what has been seen above for the strong acid, but to a lower extent (i.e., < 29.5 mV ddl−1); \({E}_{1/2}\) shifts toward more negative potentials for weak acid with pKa > 3.6 (i.e., acetic acid and dihydrogen phosphate ion), while it is almost constant for a weak acid with pKa around 3.6 (i.e., lactic acid).
The above experimental results were rationalized from a theoretical point of view by using either digital simulation or mathematical treatments [29, 37, 38, 52, 60, 68, 70,71,72]. The general case, involving both first and second-order kinetics for reaction (10), has been treated by digital simulation [37, 68, 72]. When the chemical reaction preceding the electron transfer is fast, such that the overall electrode process can proceed under diffusion control, the steady-state limiting current is given by [37]
where \({D}_{\mathrm{HA}}\) is the diffusion coefficients of HA, \({I}_{{\mathrm{l}}_{0}}\) is equal to \(4F{D}_{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}{C}_{\mathrm{A}}a\) (i.e., the limiting current which would be expected if the weak acid were fully dissociated), and \({C}_{{\mathrm{A}}^{-}}\) is the bulk concentration of A− at equilibrium. In the simulation, it is assumed that the species HA and A− share the same diffusion coefficient. \(\frac{{I}_{\mathrm{l}}}{{I}_{{\mathrm{l}}_{0}}}\) for strong acids is of course equal to 1, while for the weak acids, at the same concentration, it decreases and depends mainly on Ka. Thus, the agreement between the calculated (using Eq. (13)) and experimental \(\frac{{I}_{\mathrm{l}}}{{I}_{{\mathrm{l}}_{0}}}\) values represents a useful criterion for establishing whether the acid dissociation reaction is rapid or not. Examples of application of such criterion are displayed in Fig. 5, which compares calculated (red symbols) and experimental (black symbols) \(\frac{{I}_{\mathrm{l}}}{{I}_{{\mathrm{l}}_{0}}}\) values for series of weak acids of different Ka over a range of concentrations. The \(\frac{{I}_{\mathrm{l}}}{{I}_{{\mathrm{l}}_{0}}}\) equal to 1 for strong acids is included for reference. For weak acids with Ka > 10−5 M (i.e., monochloroacetic, lactic, and acetic), experimental and theoretical values almost overlap, indicating fast dissociation reactions. For H2PO4−, experimental \(\frac{{I}_{\mathrm{l}}}{{I}_{{\mathrm{l}}_{0}}}\) are lower than those predicted by Eq. (13) (see also the enlargement in Fig. 5) indicating that the acid dissociation step is somewhat under kinetic control. The deviation is greater at higher CA due to the circumstance that the extent of protolysis depends also on the latter parameter [1, 2]. The validity of the \(\frac{{I}_{\mathrm{l}}}{{I}_{{\mathrm{l}}_{0}}}\) criterion to identify kinetic effects on the acid dissociation reaction has also been assessed by digital simulation [37, 52, 68] using the k1 and k2 values for acetic acid and H2PO4− marked with an asterisk in Table 3. \(\frac{{I}_{\mathrm{l}}}{{I}_{{\mathrm{l}}_{0}}}\) obtained by simulation (shown in Fig. 5 with blue symbols) fit well experimental data for both acids, while those calculated by Eq. (13) fits only those for acetic acid (see inset in Fig. 5b).

\(\frac{{I}_{\mathrm{l}}}{{I}_{{\mathrm{l}}_{0}}}\) values at different CA for the acids: (1) perchloric, (2) monochloroacetic, (3) lactic, (4) acetic, (5) dihydrogen phosphate ion. Symbols refer to calculated with Eq. 13 (red), experimental (black), and simulated (blue) values. Inset: enlargement of data for acetic acid and dihydrogen phosphate ion
Another experimental way to ascertain the occurrence of the diffusion-controlled process for the weak acids is based on recording steady-state currents using microelectrodes having different radii. If the dissociation of the weak acid is fast enough to maintain equilibrium, a linear relationship between Il vs. a is found. Instead, deviation from linearity indicates the occurrence of kinetic hindering. This criterion relies on the fact that decreasing the electrode radius, the mass transport becomes more efficient (i.e., the flux rate increases) and the coupled chemical reaction is masked to an extent that depends on the size of the microelectrode [22, 27, 29, 64]. Using a range of microdisks with radii varying between about 5 and 25 µm, fast dissociation was ascertained for a series weak acid, including acetic acid, which satisfy, as anticipated above, the condition Ka/CA > 10−3 [60]. Instead, deviation from linearity is found for H2PO4− for which Ka/CA < 10−3 even for CA around 0.1 mM [60].
Considering again Eq. (13), the definition of \({I}_{{\mathrm{l}}_{0}}\), the mass balance of the acid (\({C}_{A} ={C}_{{A}^{-}}+ {C}_{\mathrm{HA}}\)), and neglecting the \({C}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}\) and \({C}_{{\mathrm{OH}}^{-}}\) due to the autoprotolysis of water (an approximation that holds for not very dilute weak acid solutions [69]), Eq. (13) can also be written in one of the following forms:
Equation (14) has also been derived by resorting to the concept of the apparent diffusion coefficient of the weak acid (Dapp) given by \({{D}_{\mathrm{app}}{C}_{\mathrm{A}}=(D}_{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}{C}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}+{ D}_{\mathrm{HA}}{C}_{\mathrm{HA}})\), which is combined with Eq. (2) [60, 68, 73]. A similar equation has also been derived analytically at hemispherical microelectrodes under the assumption that the acid dissociation step is fast [60, 71].
Equations (14) and (15) account for the lack of linearity between Il and CA, due to the circumstance that \({D}_{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}\) \(\gg\) \({D}_{\mathrm{HA}}\) (see Table 4 for DHA of a series of weak acids), and because the concentrations of the two species at equilibrium depend on \({C}_{A}\), the \(\frac{{C}_{\mathrm{HA}}}{{C}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}}\) ratio being progressively higher as CA increases [1, 2]. The Il vs. CA plots, shown in Fig. 4c with full lines for acetic and lactic acids, were calculated by Eq. (15), using the values of \({D}_{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}\) and \({D}_{\mathrm{HA}}\) indicated with an asterisk in Tables 2 and 4, the radius was set equal to 12.5 µm, and \({C}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}\) and \({C}_{\mathrm{HA}}\) were calculated by the equilibrium law, and as anticipated experimental values (symbols in Fig. 4b) agree well with the calculated ones. For \(\text{H}_2\text{PO}^-\), an apparent linearity exists due to the circumstance that both the slow acid dissociation step and the low Ka make the reduction process essentially dependent only on \({D}_{\mathrm{HA}}\) and \({C}_{\mathrm{A}}\).
The lack of linearity between Il vs. CA for most weak acids, in principle, would prevent the use of calibration plots for the detection of CA (i.e., the so-called titratable acidity [1, 2]). However, an apparent linearity can be found considering Il vs. CA plots over restricted concentration ranges, thus allowing the use of calibration plots also for the weak acids [73]. An alternative method for the detection of CA relies on the simultaneous measurements in the same acid solution of Il and pH and on the application of Eq. (15). The measured pH can be converted into \({C}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}\) (through equations T5–T6 or exploiting calibration plots of the type displayed in Fig. 3c, b), and provided that other parameters are known, CA can be calculated. This procedure avoids the need to resort to titrations curves [47], which are comparatively more time-consuming or cumbersome for some specific applications [47]. Examples of the suitability of this approach are given in Table 5 for the detection of CA of acetic and lactic acids.
Buffer solutions
Buffers formed by weak acids and their salts (i.e., the conjugate acid–base pairs) are of very practical interest, as they allow resisting to pH change of a medium on addition of an acid or base. Buffers can be obtained by mixing equal concentrations of the weak acid and its salt (CS), i.e., CA = CS, or by using different \(\frac{{C}_{\mathrm{A}}}{{C}_{\mathrm{s}}}\) ratios. The buffer concentration (CT) is given by CT = CA + CS, and the various conditions employed to prepare the buffer affect the solution pH according to the well-known Henderson − Hasselbalch equation [1, 2]:
For the special case \({C}_{{\mathrm{A}}^{-}}\)= \({C}_{\mathrm{HA}}\), pH = pKa.
The hydrogen evolution process in these systems follows paths that are essentially similar to those discussed above for the weak acid alone, as only the free hydronium and the undissociated acid at equilibrium feed the electrochemical process. Therefore, when the protolysis of HA in the buffer is fast, the CE mechanism applies and the relationships (13)–(15) can also be used to predict the steady-state diffusion limiting currents in the buffers. Furthermore, the \(\frac{{I}_{\mathrm{l}}}{{I}_{{\mathrm{l}}_{0}}}\) criterion can be employed to ascertain whether the dissociation of HA in the buffer is fast or not. As for the wave position in these systems, it depends on the acid strength and on the \(\frac{{C}_{\mathrm{A}}}{{C}_{\mathrm{s}}}\) ratio, the latter affecting in turn the pH of the medium. These aspects are highlighted below considering voltammetric data obtained with a Pt microdisk 12.5 µm radius, in a series of acetate buffers (i.e., mixtures of CH3COOH, HAc)/CH3COONa, NaAc) (Fig. 6).

Voltammetric parameters obtained in HAc/NaAc buffers, using a Pt microelectrode 12.5-µm radius. a Il vs. CA (black symbols) and pH vs. CA (blue symbols), inset \({E}_{1/2}\) vs. CA, in buffers in which \(\frac{{C}_{\mathrm{A}}}{{C}_{s}}=1\). b Il vs. CS (black symbols) and pH vs. CS (blue symbols), inset \({E}_{1/2}\) vs. CS, in buffers in which CA = 0.5 mM and CS incrementally added up to \(\frac{{C}_{\mathrm{A}}}{{C}_{\mathrm{s}}}=200\). c \(\frac{{I}_{\mathrm{l}}}{{I}_{{\mathrm{l}}_{0}}}\) vs. CS for buffers as in b; exp = experimental, eq = obtained by Eq. (14), sim = obtained by simulation
The case in which both CA and CS increase while keeping their ratio equal to 1 is of interest, as these conditions assure the highest buffer capacity [1] for a given conjugate acid-base pair. At a given CT, assuming that \({C}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}\) and \({C}_{{\mathrm{OH}}^{-}}\) are negligible compared to CA and CS, respectively, CA = \({C}_{\mathrm{HA}}\), CS = \({C}_{{\mathrm{A}}^{-}}\), and pH = pKa. Under these conditions, LSVs provide waves in which Il increases linearly (Fig. 6a, black symbols) and \({E}_{1/2}\) shifts toward more negative values (inset in Fig. 6a) by increasing CA of the buffer. Il vs. CA, over restricted concentration ranges (as those displayed in Fig. 6a), is linear, mainly because \({C}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}\) in the various buffers is almost constant, as can be verified by pH measurements using a glassy electrode (Fig. 6a, blue symbols). Therefore, also the term \({D}_{{\mathrm{H}}_{3}{\mathrm{O}}^{+}} {C}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}\) of Eq. (14) is essentially constant, and the latter equation can be rewritten in the following form:
where \(m\mathop{=}4Fa \;{D}_{{\mathrm{H}}_{3}{\mathrm{O}}^{+}} {C}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}\). The slope of the experimental Il vs. CA plot can be used to obtain the diffusion coefficient of HAc, provided that other terms are known [74, 78].
Figure 6b shows another example of buffer in which NaAc (i.e., the base conjugate to the acid) is incrementally added to a solution containing initially only CH3COOH. In this case, the steady-state limiting current decreases (Fig. 6b black symbols), while \({E}_{1/2}\) shifts toward more negative potentials (inset in Fig. 6b). This is congruent with the decrease of \({C}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}\) as is also testified by the increase of pH (Fig. 6b, blue symbols). Kinetic information on the acid dissociation reaction can be obtained by comparing experimental and theoretical \(\frac{{I}_{\mathrm{l}}}{{I}_{{\mathrm{l}}_{0}}}\). Data shown in Fig. 6c indicate that when the \(\frac{{C}_{S}}{{C}_{A}}\) ratios are large, the acid dissociation is slightly kinetically hindered. In the latter conditions \({C}_{{\mathrm{A}}^{-}}\) \(\gg\) \({C}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}\) and the backward step of reaction (10) is pseudo-first-order. These conditions are useful from a theoretical point of view, as they allow simplifying the mathematical treatment of the diffusion problem for the evaluation analytically of k2, and through the relationship Ka = k1/k2, also k1 of the weak acid. An example of this treatment is reported in Ref. [64]. The differential equations for the concentrations of the various species involved in the reaction have been solved in a spherical diffusion field for HAc/NaAc buffers and using CA and CS such that the conditions \({D}_{\mathrm{HA}}{C}_{\mathrm{HA}}\) \(\gg\) \({D}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}_{ }} {C}_{{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}_{ }}}\) and \({k}_{2}{C}_{{\mathrm{A}}^{-}}{D}_{\mathrm{HA}}\gg\) \({k}_{1}{C}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}\) apply. Under these conditions, the following analytical solution for the steady-state current density (il) can be obtained [64]:
where \({r}_{\mathrm{s}}\) is radius of a microsphere. This equation can also be used for a microdisk, considering that the current density at the microdisk is identical to that of a microsphere of radius \({r}_{\mathrm{s}}= \frac{\pi a}{4}\) [27, 64, 79, 80]. Equation (18) for the microdisk can therefore be rewritten as follows:
The plot of \(\frac{1}{{I}_{\mathrm{l}}}\) vs. a provides a straight line of slope (S) and intercept (Y), given by the following relationships:
From the slope, \({C}_{\mathrm{HA}}\) can be evaluated. In addition, considering that \({k}_{1}{C}_{\mathrm{HA}}={k}_{2}{C}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}_{ }} {C}_{{\mathrm{A}}^{-}}\) and substituting k1 in Y, one obtains
\({C}_{{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}}\) can be obtained by using Ka. In his way k2 equal to 4.1 × 10 M s−1 and 4.3 × 1010 M s−1 in two buffers made of 40 mM HAc/1 M NaAc and 70 mM HAc/1 M NaAc, respectively, were evaluated [64]. It must be considered that for these measurements, it is necessary to have available a range of microelectrodes of different radii.
Mixtures of monoprotic, polyprotic acids and buffer solutions
The above treatments can be extended to mixtures of monoprotic, polyprotic acids and buffers formed by using a variety of acid–base pairs. For these systems, the hydrogen evolution process results from the simultaneous protolysis equilibria of the type (10), each due to donating protons of the acids, coupled with reaction (11). Steady-state limiting currents, half-wave potentials, and other voltammetric parameters depend on the acidity constant, kinetics of each equilibrium, concentrations of the species at equilibrium, and their diffusivities. In aqueous solutions, in general, single or at most two waves can be distinguished, each of them including one or more species, depending on the complexity of the mixture. This has been observed using either LSV or square-wave voltammetry (SWV), the latter allowing to improve the separation of the waves [61, 81, 82]. Using LSV and considering the additivity of currents that applies for steady-state voltammograms, an equation of the type (15) can be derived for any mixture of acids, provided that all dissociation steps are fast. The equation links the overall steady-state limiting current to the overall analytical concentration of all acids in the mixture. Thus, as noticed above, the simultaneous measurements of steady-state limiting current and pH allow obtaining the titratable acidity of the medium. Readers are referred to the cited literature [47, 52, 60,61,62, 78] for more detailed accounts of these systems.
Oxygen evolution from strong and weak bases at gold microelectrodes
The oxygen evolution process, recorded in relatively dilute aqueous solutions of both strong and weak bases, parallels, to some extent, that described above for the hydrogen evolution reaction recorded in acid solutions. Therefore, under given conditions, theoretical relationships and explanations similar to those given above can be used/invoked to rationalize the general voltammetric behavior of bases. For the latter measurements, gold microelectrodes proved more suitable [83, 84], compared, for example, to platinum and nickel microelectrodes, which could also be used for the same purposes [85, 86]. In a few reports, boron-doped diamond [87], mesoporous titanium dioxide materials [88], tungsten-based materials [89], and platinum-modified zeolite electrode [90] have been used as efficient material for OH− oxidation. In all cases, the main electrode reaction for strong bases involves the direct oxidation of hydroxide ions:
producing one electron for each OH− ion, with a nonunity stoichiometry. For weak bases, reaction (23) is preceded by the chemical step:
characterized by the basicity constant Kb (Table 1) and the forward (kf) and backward (kb) rate constants. In what follows, examples exploiting the above reactions are limited to the strong base NaOH and the weak base NH3.
Figure 7 (main picture) shows typical LSVs obtained at an Au microdisk 12.5-µm radius in a solution containing 1.5 mM of NaOH or NH4OH (NH3 +H2O).

LSV responses recorded at a Au microelectrode, 12.5-µm radius, in 0.1 M NaClO4 aqueous solutions containing 1.70 mM NH3 (blue line) and 1.5 mM NaOH (red line), scan rate 5 mVs−1. Inset: Emid vs. a (open symbols) and concentration C (filled symbols) plots for NH3 (blue symbols) and NaOH (red symbols) (figure adapted from Ref. [91] with permission)
Well-defined waves at potentials above 1.2 V vs. Ag/AgCl are observed in both cases; the steady-state limiting currents decrease and the potentials at half the diffusion limiting current (Emid, not strictly equal to \({E}_{1/2}\), as the latter refers to reversible processes [92]) depend on the base strength, as well as on the radius of the microelectrode employed. At a given analytical concentration of the base (CB), the steady-state limiting current decreases (main picture) and Emid shifts toward more positive values as pKb or the microdisk radius decrease (inset in Fig. 7, filled symbols). This behavior is related to the fact that reaction (23), unlike reaction (11), is irreversible [83, 85, 92, 93], due to the formation of gold oxides onto the electrode surface, which impose a barrier to the heterogeneous charge transfer at the electrode/electrolyte interface [83, 85, 94]. In these measurements, the use of low scan rates (i.e., 5–10 mV s−1) is mandatory, as at larger scan rates a broad wave at much less positive potentials (at about 0.6 V in Fig. 8), due oxide formation, becomes evident. Correspondingly, the wave at 1.4 V (Fig. 8) is much less defined and difficult to analyze. Instead, at low scan rates, the waves recorded in either NaOH or NH3 solutions, using gold microdisks with different radii (over the range 2.5–30 µm), showed good linearity between Il vs. a, indicating that the OH− oxidation is diffusion controlled [83, 85, 91, 93].

LSV responses recorded at a Au microelectrode, 12.5 µm radius, in a 0.1 M Na2SO4 aerated aqueous solution containing 1.0 mM NaOH at scan rate: 5 (blue line), 50 (cyan line), 100 (pink line), and 200 mV s−1 (green line)
The analytical concentration of the base affects the LSV profiles, and this also depends on the base strength. Figure 9a (main picture) shows a series of LSVs recoded at a 12.5-µm radius Au microdisk in aqueous solutions containing NaOH in the concentration range 0.1–3 mM and 0.1 M Na2SO4 as supporting electrolyte. Il increases with the increase of the base concentration, and it can be predicted by [83, 84, 93]
where \({D}_{{\mathrm{OH}}^{-}}\) and \({C}_{{\mathrm{OH}}^{-}}\) are the diffusion coefficient and bulk concentration of the hydroxide ion, respectively. Since \({C}_{{\mathrm{OH}}^{-}}= {C}_{\mathrm{B}}\), linearity exists between Il and CB (Fig. 9b). From the slope of the plot, \({D}_{{\mathrm{OH}}^{-}}\) can be evaluated; the value obtained from data of Fig. 9b is shown in Table 6 [93], which compares well with others available in the literature (Table 6).

a LSV responses recorded at a Au microelectrode, 12.5-µm radius, in 0.1 M Na2SO4 aqueous solutions containing NaOH at increasing concentrations: 0.2 (red line), 0.65 (black line), 1.25 (green line), and 2.8 mM (blue line); scan rate 5 mVs−1. Inset: LSV responses recorded at the same Au microdisk and scan rate, in 0.1 M Na2SO4 and 30 mM NaOH (with permission from Ref. [85]). b Il vs. CB plot. c log Il vs. pH plot
As for the wave position, Emid shifts toward more positive values by increasing CB, as higher OH− concentrations lead to the formation of thicker gold oxide layers. This in turn leads to further inhibition of the electron transfer process, to such an extent that the oxidation wave even splits (inset in Fig. 9a). However, the overall steady-state limiting current recorded at higher overpotentials (i.e., at about 1. 6 V vs. SCE, in Fig. 9a) still depends linearly on the analytical concentration of the base [85].
The steady-state limiting current is related to pH through equations T7–T9:
where symbols have their usual meaning. Figure 9c shows experimental log Il vs. pH plot obtained from the simultaneous LSV and pH measurements in aqueous solutions containing NaOH at different concentrations and 0.1 M Na2SO4 as supporting electrolyte. The experimental plot displays a slope equal to 1.02, close to theoretical value predicted by Eq. (26).
For a weak base with no very low Kb, such that the kinetic of reaction (20) is fast enough to support the mass transfer control, the steady-state limiting current can be predicted by an equation similar to (14). This is the case of NH3 (pKb = 3.02 × 10−5 at 0.1 M ionic strength [103] or 1.75 × 10−5 at 0 ionic strength), which in aqueous solution undergoes the following acid–base equilibrium:
The forward and backward rate constants kf = 5.0 × 105 s-1and kb = 3 × 1010 M−1 s−1 [104] and kf 8.3 × 105 s−1 and kb = 2.8 × 1010 M−1 s−1 [91] ensure mass transport control. Therefore, the steady-state limiting current is given by [91]
where \({D}_{{\mathrm{NH}}_{3}}\) and \({C}_{{\mathrm{NH}}_{3}}\) are the diffusion coefficient and bulk concentration at equilibrium of ammonia. In the derivation of the latter equation, it is also assumed that NH3 and NH4+ share the same diffusion coefficient. Since \({D}_{{\mathrm{OH}}^{-}}\) > \({D}_{{\mathrm{NH}}_{3}}\) (see Table 5), also for weak base, no linearity should exist over a wide concentration range. However, in this case, the difference between \({D}_{{\mathrm{OH}}^{-}}\) and \({D}_{{\mathrm{NH}}_{3}}\) is smaller than in the case of \({D}_{{\mathrm{H}}_{3}{\mathrm{O}}^{+}}\) and \({D}_{\mathrm{HA}}\); therefore, deviation from linearity mainly applies in the lower CB range, where the base dissociation is relatively larger.
The \(\frac{{I}_{\mathrm{l}}}{{I}_{{\mathrm{l}}_{0}}}\) criterion can be used to establish whether the chemical reaction is fast enough to support diffusion-controlled currents. In this case, \({I}_{{\mathrm{l}}_{0}}\) corresponds to the steady state limiting current of the weak base assumed to be completely dissociated. The validity of Eq. (28) was confirmed by digital simulation. Figure 10 compares calculated by Eq. (28), simulated and experimental Il vs. CB values recorded at two gold microdisks of 13 and 5 µm radius in solutions containing NH3 over the concentration range 0.1–10 mM.

 ) Eq. (
) Eq. ( ) simulation, and (
) simulation, and (
 ) experimental values (adapted from Ref. [
) experimental values (adapted from Ref. [Voltammograms like those described above for the weak base can also be obtained in buffer solutions obtained by mixtures of the weak base and the salts containing the conjugate acid. As examples, the case in which NH3 is mixed with either different or equal amounts of NH4ClO4 or NH4NO3 [91] is mentioned. LSVs having a shape as those shown in Fig. 7 (blue line) can be recorded. Moreover, the steady-state limiting current is proportional to the analytical concentration of ammonia of the buffer when it was kept over the range 1–10 mM, provided that the OH− concentration (and therefore the pH of the medium) remains constant [91]. For instance, it was shown that in a 10 mM NH4ClO4 solution using a 5-μm radius gold microdisk, the steady-state limiting current was proportional to the analytical concentration of ammonia, indicating that NH3 is the diffusion species that mainly feeds the electrode process [91].
Conclusions
In this article, we have highlighted some experimental and theoretical aspects on how acid–base equilibria can be investigated by voltammetry at microelectrodes, exploiting the hydrogen and oxygen evolution processes. The advantages of using microelectrode voltammetry relies mainly in the attainment of reproducible and well-defined convergent mass-transport conditions and the achievement of a steady-state diffusion regime in short times. This enables the use of a one-dimensional hemispherical approximation [52, 64] to derive analytical solutions to predict voltammetric responses. However, most information provided in this article has been modeled by the two-dimensional simulations of the investigated systems. Although we did not enter details on these methods, we hope that the illustrated examples will arouse the interest of teachers and students toward modeling electrochemical processes using mathematical and simulation methods, generally overlooked in electrochemical curricula. We noted that some articles, included in this Education & Electrochemistry collection, deal with the use simulations in electrochemistry for educational curricula [105], or highlight the usefulness of the use of computer or simulations to learn various concepts inherent the electrochemical experiments [106]. In particular, in Ref. [106], the author highlights the state of the art in computer and simulations in electrochemistry curricula and the benefit of existing computer-aided methods and techniques to improve students’ ability to learn various concepts inherent in the electrochemical experiments. We agree with the authors’ analysis, from which it appears that these aspects are overlooked in the education of electrochemistry [106]. Therefore, it is hoped that considerable effort will be made by the electrochemical community to adopt appropriate educational programs that include computer-aided approaches. Commercial software such as KISSA and DIGISIM (currently DigiElch) Comsol and open-source electrochemistry simulation packages [107,108,109], together with educational examples, are available to study mechanisms and extract kinetic parameters of electrode processes. Besides, the more recent machine learning (or artificial intelligence) methods, which have emerged as a powerful tool in electrode mechanism classification and parametrization, would also be useful, as it accelerates the prediction of entire voltammetric responses on the basis of known parameters (i.e., kinetic and thermodynamic constants, formal potential, and diffusion coefficients) or vice versa [70, 110]. For instance, artificial intelligence has been applied to predict thermodynamics and kinetics of the dissociation of acetic acid in aqueous solution [70], as well as the current vs. potential profile, with good agreement with experimental results.
Finally, we would like to point out that the methods described in this article to study and quantify acids and bases in aqueous solutions can be extended to other nonaqueous protic solvents (e.g., methanol and ethanol), aprotic solvents (e.g., acetonitrile and DMSO), and room temperature ionic liquids (RTILs). The chemical and physical nature of the solvents of course plays a role as they affect the acid–base equilibria and the strength of acids and bases. These aspects have not been considered here. Articles reporting investigations on acids and bases in nonaqueous solvents performed by either macro or microelectrode voltammetry are available. Readers interested in such topics may refer to the literature cited and references included therein [111,112,113,114,115,116,117,118,119,120,121,122].
References
Butler JN, Cogley DR (1998) Ionic equilibrium: solubility and pH calculations. Wiley, New York
Scholz F (2015) Voltammetric techniques of analysis: the essentials. ChemTexts 1. https://doi.org/10.1007/978-3-030-17180-3
Eknoyan G (2022) Acid–base homeostasis: a historical inquiry of its origins and conceptual evolution. Nephrol Dial Transplant 37:1816–1823. https://doi.org/10.1093/ndt/gfac014
Lücke FK, Adams MR (2023) Chapter 22 - acids and fermentation. In: Andersen V, Lelieveld H, Motarjemi Y (eds) Food safety management, Second Edition. Academic Press, San Diego, pp 439–452. https://doi.org/10.1016/B978-0-12-820013-1.00020-6
Garsany Y, Pletcher D, Hedges B (2002) Speciation and electrochemistry of brines containing acetate ion and carbon dioxide. J Electroanal Chem 538–539:285–297. https://doi.org/10.1016/S0022-0728(02)00728-3
Salarirada MM, Behnamfardb A, Veglio F (2021) Desalination and water treatment. Removal of xanthate from aqueous solutions by adsorption onto untreated and acid/base treated activated carbons. Desalin Eater Treat 212:220–223. https://doi.org/10.5004/dwt.2021.26683
Domínguez JR, Durán-Valle CJ, Mateos-García G (2022) Synthesis and characterisation of acid/basic modified adsorbents. Application for chlorophenols removal. Environ Res 207:112187. https://doi.org/10.1016/j.envres.2021.112187
Kassier JP (1982) Historical perspectives In: Cohen JJ, Kassier JP (eds) Acid-base. Little Brown, Boston
Hildebrand JH (1981) A history of solution theory. Ann Rev Phys Chem 32:1–24
Arrhenius S (1903) Development of the theory of electrolyte dissociation. Nobel Lectures 11 December 1903. https://www.nobelprize.org/prizes/chemistry/1903/arrhenius/lecture/. Accessed 20 Jun 2023
Malkin HM (2003) Historical review: concept of acid-base balance in medicine. Ann Clin Lab Sci 33:337–344
Kauffman GBJ (1988) The Brønsted-Lowry acid-base concept Chem Educ 59:28–31
Brønsted JN (1923) Some remarks on the concept of acids and bases. Recl Trav Chim Pays-Bas 42:718–728
Jensen WB (1980) The Lewis acid-base concepts: an overview. Wiley, New York. http://www.scribd.com/doc/52094935/The-Lewis-Acid-Base-Concepts-William-B-Jensen#scribd
McNaught AD (1997) IUPAC. Compendium of chemical terminology (the “Gold Book”), 2nd edn. Blackwell Scientific Publications, Oxford. https://doi.org/10.1351/goldbook
Bell RP (1959) The hydronium in chemistry. Methuen, London
Kolthoff IM, Elving PJ (1961) Treatise on analytical chemistry, 2nd edn. Interscience Encyclopedia, New York
Aroti A, Leontidis E (2001) Simultaneous determination of the ionization constant and the solubility of sparingly soluble drug substances. A physical chemistry experiment. J Chem Educ 78:786. https://doi.org/10.1021/ed078p786
Nyasulu F, McMills L, Barlag R (2013) Weak acid ionization constants and the determination of weak acid–weak base reaction equilibrium constants in the general chemistry laboratory. J Chem Educ 90:768–770. https://doi.org/10.1021/ed300403v
Yimkosol W, Dangkulwanich M (2021) Finding the pKa values of a double-range indicator thymol blue in a remote learning activity. J Chem Educ 98:3930–3934. https://doi.org/10.1021/acs.jchemed.1c00122
Heyrovsky J, Kuta J (1965) Principles of polarography. Prague. https://doi.org/10.1016/C2013-0-10851-3
Bard AJ, Faulkner LR (2001) Electrochemical methods: fundamentals and applications, 2nd edn. Wiley, New York, USA
Opekar F, Beran P (1976) Rotating disk electrodes. J Electroanal Chem Interfacial Electrochem 69:1–105. https://doi.org/10.1016/S0022-0728(76)80129-5
Vetter KJ (1967) Electrochemical kinetics-theoretical and experimental aspects. In: Academic Press (ed). New York
Stulík K, Amatore C, Holub K, Marecek V, Kutner W (2000) Microelectrodes. Definitions, characterization, and applications. Pure Appl Chem 72:1483–1492. https://doi.org/10.1351/pac200072081483
Daniele S, Denuault G (2014) From microelectrodes to scanning electrochemical microscopy. In: Pletcher D, Tian Z-Q, Williams DE (eds) Developments in electrochemistry. Science inspired by Martin Fleischmann. Wiley, New York, USA, pp 223–244. https://doi.org/10.1002/9781118694404.ch12
Montenegro MI, Queirós MA, Daschbach JL (1991) Microelectrodes: theory and applications. NATO ASI Ser E Appl Sci 197. Kluwer, Dordrecht, The Netherlands. https://doi.org/10.1007/978-94-011-3210-7
Wightman RM, Wipf DO (1989) Voltammetry at ultramicroelectrodes. In: Bard AJ (ed) Electroanalytical chemistry. A series of advances, vol 15. Marcel Dekker, New York, USA, pp 267–344
Fleischmann M, Pons S, Rolison DR, Schmidt PP (1987) Ultramicroelectrodes. Datatech Systems and Technology Inc, Morganton, NC, USA
Wang J (2023) Analytical electrochemistry. In: Sons, John Willey & Sons I (ed). Hoboken, NJ, USA, p 240
Scholz F (2015) Voltammetric techniques of analysis: the essentials. ChemTexts 1:17. https://doi.org/10.1007/s40828-015-0016-y
Buck RP, Rondinini S, Covington AK, Baucke FGK, Brett CMA, Camoes MF, Milton MJT, Mussini T, Naumann R, Pratt KW, Spitzer P, Wilson GS (2002) Measurement of pH. Definition, standards, and procedures (IUPAC Recommendations 2002). Pure Appl Chem 74:2169–2200
Compton RG, Banks CE (2018) Understanding voltammetry, 3rd edn. Imperial College Press, London
Newell DB, Tiesinga E (2019) The International System of Units (SI). NIST Special Publication, Gaithersburg, Maryland. https://doi.org/10.6028/NIST.SP.330-2019
Saito Y (1968) A theoretical study on the diffusion current at the stationary electrodes of circular and narrow band types. Rev Polarog 15:177–187. https://doi.org/10.5189/revpolarography.15.177
Daniele S, Bragato C (2014) From macroelectrodes to microelectrodes. Theory and properties. In: Environmental analysis by electrochemical sensors and biosensors. Springer New York
Daniele S, Lavagnini I, Baldo MA, Magno F (1996) Steady state voltammetry at microelectrodes for the hydrogen evolution from strong and weak acids under pseudo-first and second order kinetic conditions. J Electroanal Chem 404:105–111. https://doi.org/10.1016/0022-0728(95)04348-9
Jaworski A, Donten M, Stojek Z, Osteryoung JG (1999) Conditions of strict voltammetric reversibility of the h(+)/h(2) couple at platinum electrodes. Anal Chem 71:243–246. https://doi.org/10.1021/ac9804240
Jiao X, Batchelor-McAuley C, Kätelhön E, Ellison J, Tschulik K, Compton RG (2015) The subtleties of the reversible hydrogen evolution reaction arising from the nonunity stoichiometry. J Phys Chem C 119:9402–9410. https://doi.org/10.1021/acs.jpcc.5b01864
Qin SF, Cheng FF, Le-Xing You LX, Jian-Jun Sun JJ (2022) Fundamentals on kinetics of hydrogen redox reaction at a polycrystalline platinum disk electrode. J Phys Chem 922:116788. https://doi.org/10.1016/j.jelechem.2022.116788
Zhou M, Bao S, Bard AJ (2019) Probing size and substrate effects on the hydrogen evolution reaction by single isolated Pt atoms, atomic clusters, and nanoparticles. J Am Chem Soc 141:7327–7332. https://doi.org/10.1021/jacs.8b13366
Macpherson JV, Unwin PR (1997) Determination of the diffusion coefficient of hydrogen in aqueous solution using single and double potential step chronoamperometry at a disk ultramicroelectrode. Anal Chem 69:2063–2069. https://doi.org/10.1021/ac961211i
Ciszkowska M, Stojek Z, Morris SE, Osteryoung JG (1992) Steady-state voltammetry of strong and weak acids with and without supporting electrolyte. Anal Chem 64:2372–2377. https://doi.org/10.1021/ac00044a013
Bockris JO'M, Reddy AKN (1970) Modern electrochemistry. In: Modern electrochemistry. New York, pp 381–387
Bockris JO'M, Reddy AKN (1970) Modern electrochemistry. In: Modern electrochemistry. New York, pp 550–552
Daniele S, Ugo P, Mazzochin GA, Rudello D (1992) Effects of dispersed phases in naturally occurring fluids on the electroreduction of Ru(NH3)6Cl3. Mackay RA and Texter J Eds. VCH, New York
Daniele S, Bragato C, Baldo MA, Mori G, Giannetto M (2001) A novel approach for the determination of the total concentration of acids in aqueous solutions by simultaneous diffusion limited current for reduction of acids and pH measurements. Anal Chim Acta 432:27–37. https://doi.org/10.1016/S0003-2670(00)01362-3
Stackelberg MV, Pilgram M (1960) Diffusionskoeffizient des wasserstoffions in wässrigen KCl-Lösungen 25:2974–2976
Lanning JA, Chambers JQ (1973) Voltammetric study of the hydrogen ion/hydrogen couple in acetonitrile/water mixtures. Anal Chem 45:1010–1016. https://doi.org/10.1021/ac60329a002
Chen Q, Luo L (2018) Correlation between gas bubble formation and hydrogen evolution reaction kinetics at nanoelectrodes. Langmuir 34:4554–4559. https://doi.org/10.1021/acs.langmuir.8b00435
Zhou J, Zu Y, Bard AJ (2000) Scanning electrochemical microscopy: part 39. The hydronium/hydrogen mediator system and its application to the study of the electrocatalysis of hydrogen oxidation. J Electroanal Chem 491:22–29. https://doi.org/10.1016/S0022-0728(00)00100-5
Jaworski A, Donten M, Stojek Z (1999) Migration and diffusion coupled with a fast preceding reaction. Voltammetry at a microelectrode. Anal Chem 71(1):167–173
Wetsema BJC, Mom HJM, Los JM (1976) Pulse polarography: IX. Hydronium diffusion in aqueous sulphate solution. J Electroanal Chem Interfacial Electrochem 68:139–147. https://doi.org/10.1016/S0022-0728(76)80002-2
Lee SH, Rasaiah JC (2011) Hydronium transfer and the mobilities of the H+ and OH− ions from studies of a dissociating model for water. J Chem Phys 135:124505. https://doi.org/10.1063/1.3632990
Tomeš J (1937) Polarographic studies with the dropping mercury kathode. LXIII. Verification of the equation of the polarographic wave in the reversible electrodeposition of free kations. Collect Czechoslov Chem Commun 9:12–21. https://doi.org/10.1135/cccc19370012
Bond AM, Oldham KB, Zoski CG (1989) Steady-state voltammetry. Anal Chim Acta 216:177–230. https://doi.org/10.1016/S0003-2670(00)82009-7
Gómez-Gil JM, Laborda E, Molina A (2020) General explicit mathematical solution for the voltammetry of nonunity stoichiometry electrode reactions: diagnosis criteria in cyclic voltammetry. Anal Chem 92:3728–3734. https://doi.org/10.1021/acs.analchem.9b05023
González J, Laborda E, Serna C, Torralba E, Molina A (2021) Steady state voltammetry of charge transfer processes with nonunity electrode reaction orders. J Electroanal Chem 896:115206. https://doi.org/10.1016/j.jelechem.2021.115206
Nürnberg HW (1964) Proceedings of the third international congress of polarography. Southampton, pp 149–186
Stojek Z, Ciszkowska M, Osteryoung JG (1994) Self-enhancement of voltammetric waves of weak acids in the absence of supporting electrolyte. Anal Chem 66:1507–1512. https://doi.org/10.1021/ac00081a024
Bragato C, Daniele S, Baldo MA (2005) Low frequency square-wave voltammetry of weak acids at platinum microelectrodes. Electroanalysis 17:1370–1378. https://doi.org/10.1002/elan.200503281
Ciszkowska M, Stojek Z, Osteryong JG (1995) Voltammetric reduction of polyprotic acids at the platinum microelectrode: dependence on supporting electrolyte. J Electroanal Chem 398:49–56. https://doi.org/10.1016/0022-0728(95)04246-X
Kanzaki Y, Tokuda K, Bruckenstein S (2014) Dissociation rates of weak acids using sinusoidal hydrodynamic modulated rotating disk electrode employing Koutecky-Levich equation. J Electrochem Soc 161:H770–H779. https://doi.org/10.1149/2.0221412jes
Fleischmann M, Lasserre F, Robinson J, Swan D (1984) The application of microelectrodes to the study of homogeneous processes coupled to electrode reactions: part I. EC′ and CE reactions. J Electroanal Chem Interfacial Electrochem 177:97–114. https://doi.org/10.1016/0022-0728(84)80215-6
Nürnberg HW, Dürbeck HW, Wolff G (1967) Die voltammetrische bestimmung der geschwindigkeitskonstanten der dissoziation und rekombination einiger carbonsäuren mit strukturell bedingten abweichungen vom normalverhalten. Z Phys Chem 52:144–169. https://doi.org/10.1524/zpch.1967.52.1_4.144
Eigen M, Eyring EM (1962) Fast protolytic reactions in aqueous solutions of aminobenzoic acids. J Am Chem Soc 84:3254–3256. https://doi.org/10.1021/ja00876a008
Staples BR, Turner DJ, Atkinson G (1969) An apparatus for the determination of rates of hydronium transfer and other very fast reactions. Instrum Sci Technol 2:127–147. https://doi.org/10.1080/10739146908543273
Daniele S, Lavagnini I, Baldo MA, Magno F (1998) Voltammetry for reduction of hydrogen ions from mixtures of mono- and polyprotic acids at platinum microelectrodes. Anal Chem 70:285–294. https://doi.org/10.1021/ac970666k
Albery WJ (1966) Effect of the dissociation of water on electrochemical studies involving hydrogen ions. Trans Faraday Soc 62:1575–1582. https://doi.org/10.1039/TF9666201575
Chen H, Kätelhön E, Haonan L, Compton RG (2021) Use of artificial intelligence in electrode reaction mechanism studies: predicting voltammograms and analyzing the dissociative CE Reaction at a hemispherical electrode. Anal Chem 93:13360–13372. https://doi.org/10.1021/acs.analchem.1c03154
Oldham KB (1996) Effect of diffusion coefficient diversity on steady-state voltammetry when homogeneous equilibria and migration are encountered. Anal Chem 68:4173–4179. https://doi.org/10.1021/ac9601730
Jaworski A, Stojek Z, Osteryoung JG (1995) A simple theoretical model for the self-enhancement of the cathodic voltammetric waves of weak acids. Anal Chem 67:3349–3352. https://doi.org/10.1021/ac00114a035
Daniele S, Baldo MA, Simonetto F (1996) Assessment of linearity between steady-state limiting current and analytical concentration of weak acids in the reaction of hydrogen evolution. Anal Chim Acta 331:117–123. https://doi.org/10.1016/0003-2670(96)00188-2
Canhoto C, Matos M, Rodrigues A, Geraldo MD, Bento MF (2004) Voltammetric analysis of weak acids with microelectrodes. J Electroanal Chem 570:63–67. https://doi.org/10.1016/j.jelechem.2004.03.015
Li T, Xu J, Zhou L, Wang L, Sun Y, Cheng J, Yuan M, Wang L, Yue Y, Wang J (2013) Voltammetric determination of the adsorption kinetics of acetic acid on activated carbon. J Electrochem Soc 160:H568. https://doi.org/10.1149/2.033309jes
Lide DR (2010) CRC handbook of chemistry and physics, 88th edn. CRC Press/Taylor&Francis Group, Boca Raton, FL, USA
Burkell JE, Spinks JWT (1952) Measurements of self-diffusion in aqueous solutions of sodium dihydrogen phosphate. Can J Chem 30:311–319. https://doi.org/10.1139/v52-042
Daniele S, Baldo MA, Bragato C, Lavagnini I (1998) Steady state voltammetry in the process of hydrogen evolution in buffer solutions. Anal Chim Acta 361:141–150. https://doi.org/10.1016/S0003-2670(97)00695-8
Oldham KB (1981) Edge effects in semiinfinite diffusion. J Electroanal Chem Interfacial Electrochem 122:1–17. https://doi.org/10.1016/S0022-0728(81)80136-2
Heinze J (1981) Diffusion processes at finite (micro) disk electrodes solved by digital simulation. J Electroanal Chem Interfacial Electrochem 124:73–86. https://doi.org/10.1016/S0022-0728(81)80285-9
Matos M, Canhoto C, Bento MF, Geraldo MD (2010) Simultaneous evaluation of the dissociated and undissociated acid concentrations by square wave voltammetry using microelectrodes. J Electroanal Chem 647:144–149. https://doi.org/10.1016/j.jelechem.2010.06.010
Daniele S, Bragato C, Baldo MA (2002) Square wave voltammetry of strong acids at platinum microelectrodes. Electrochem commun 4:374–378. https://doi.org/10.1016/S1388-2481(02)00320-X
Abdelsalam ME, Denuault G, Baldo MA, Daniele S (1998) Voltammetry of hydroxide ion in aqueous solutions on gold microelectrodes. J Electroanal Chem 449:5–7. https://doi.org/10.1016/S0022-0728(98)00053-9
Ordeig O, Banks C, Davies T, Del Campo F, Muñoz F, Compton R (2006) Gold ultra-microelectrode arrays: application to the steady-state voltammetry of hydroxide ion in aqueous solution. Anal Sci 22:679–683. https://doi.org/10.2116/analsci.22.679
Abdelsalam ME, Denuault G, Baldo MA, Bragato C, Daniele S (2001) Detection of hydroxide ions in aqueous solutions by steady-state voltammetry. Electroanalysis 13:289–294. https://doi.org/10.1002/1521-4109(200103)13:4%3c289::AID-ELAN289%3e3.0.CO;2-F
Ciani I, Daniele S (2004) Oxidation of hydroxide ions from dilute unbuffered solutions of ammonia at platinum microdiscs surrounded by thick and thin insulating sheaths. J Electroanal Chem 564:133–140. https://doi.org/10.1016/j.jelechem.2003.09.032
Irkham EY (2019) Oxidation of hydroxide ions in weak basic solutions using boron-doped diamond electrodes: effect of the buffer capacity. Analyst 144:4499–4504. https://doi.org/10.1039/C9AN00505F
Amer MS, Ghanem MA, Al-Mayouf AM (2020) Hydroxide ion oxidation using low-symmetry mesoporous titanium dioxide (lsm-TiO2) electrode. J Electroanal Chem 871:114268. https://doi.org/10.1016/j.jelechem.2020.114268
Pašti I, Lazarević-Pašti T, Mentus S (2012) Switching between voltammetry and potentiometry in order to determine H+ or OH− ion concentration over the entire pH scale by means of tungsten disk electrode. J Electroanal Chem 665:83–89. https://doi.org/10.1016/j.jelechem.2011.11.019
Mudrinić T, Mojović Z, Rabi-Stanković A, Ivanović-Šašić A, Milutinović-Nikolić A, Jovanović D (2012) Oxidation of hydroxide ions at platinum-modified zeolite electrode. Hem Ind 66:759–767. https://doi.org/10.2298/HEMIND111223027M
Daniele S, Baldo MA, Bragato C, Abdelsalam ME, Denuault G (2002) Steady-state voltammetry of hydroxide ion oxidation in aqueous solutions containing ammonia. Anal Chem 74:3290–3296. https://doi.org/10.1021/ac025530n
González J, Laborda E, Molina Á (2023) Voltammetric kinetic studies of electrode reactions: guidelines for detailed understanding of their fundamentals. J Chem Educ 100:697–706. https://doi.org/10.1021/acs.jchemed.2c00944
Daniele S, Baldo MA, Bragato C, Denuault G, Abdelsalam ME (1999) Steady-state voltammetry for hydroxide ion oxidation in aqueous solutions in the absence of and with varying concentrations of supporting electrolyte. Anal Chem 71:811–818. https://doi.org/10.1021/ac9807619
Abu-Rabi A, Jašin D, Mentus S (2007) The influence of cathodic pretreatment on the kinetics of hydroxide ion oxidation on polycrystalline gold electrode. J Electroanal Chem 600:364–368. https://doi.org/10.1016/j.jelechem.2006.09.009
Wiberg GKH, Arenz M (2015) On the influence of hydronium and hydroxide ion diffusion on the hydrogen and oxygen evolution reactions in aqueous media. Electrochim Acta 159:66–70. https://doi.org/10.1016/j.electacta.2015.01.098
Breiter M, Hoffmann K (1960) Bestimmung der diffusions koeffizienten DHs, D, und DOH-mit der platinscheibenelektrode. Z Elektrochem 64:462
Atkins P, Paula JD (2002) Physical chemistry, 7th edn. WH Freeman and Company, New York, USA
Littauer EL, Tsai KC (1979) Observations of the diffusion coefficient of the hydroxyl ion in lithium hydroxide solutions. Electrochim Acta 24:351–355. https://doi.org/10.1016/0013-4686(79)87018-8
Lopez de Mishima BA, Lescano DT, Molina Holgado HT (1998) Electrochemical oxidation of ammonia in alkaline solutions: its application to an amperometric sensor. Electrochim Acta 43:395–404. https://doi.org/10.1016/S0013-4686(97)00061-3
Donten M, Hyk W, Wojciech H, Ciszkowska M, Stojek Z (1997) Electrooxidation of ammonia and simple amines at titanium electrodes modified with a mixture of ruthenium and titanium dioxides. Electroanalysis 9:751–754. https://doi.org/10.1002/elan.1140091004
Donten M, Osteryoung J (1991) Electrochemical and chemical reactions in baths for plating amorphous alloys. J Appl Electrochem 21:496–503. https://doi.org/10.1007/BF01018601
International critical tables of numerical data, physics, chemistry, and technology (1930) National Research Council of the United States of America, Washington DC, USA. https://doi.org/10.17226/20230
Martell AE, Smith RM (1981) Critical stability constants. Plenum Press, New York
Eigen M, Schoen J (1955) Potential-impulse method for the investigation of very rapid ionic reactions in aqueous solution. J Z Elektrochem 59:483–494
Gulaboski RMV (2023) Calculating of square-wave voltammograms-a practical on-line simulation platform. J Solid State Electrochem. https://doi.org/10.1007/s10008-023-05520-y
Bieniasz LK (2023) While educating electrochemists, do not forget we live in a computer era. J Solid State Electrochem. https://doi.org/10.1007/s10008-023-05457-2
Wang X, Wang Z (2022) Animated electrochemistry simulation modules. J Chem Educ 99:752–758. https://doi.org/10.1021/acs.jchemed.1c00944
Wang S, Wang J, Gao Y (2017) Development and use of an open-source, user-friendly package to simulate voltammetry experiments. J Chem Educ 94:1567–1570. https://doi.org/10.1021/acs.jchemed.6b00986
Khalafi L, Cunningham AM, Hoober-Burkhardt LE, Rafiee M (2021) Why is voltammetric current scan rate dependent? Representation of a mathematically dense concept using conceptual thinking. J Chem Educ 98:3957–3961. https://doi.org/10.1021/acs.jchemed.1c00770
Bond AM, Zhang J, Gundry L, Kennedy GF (2022) Opportunities and challenges in applying machine learning to voltammetric mechanistic studies. Curr Opin Electrochem 34:101009. https://doi.org/10.1016/j.coelec.2022.101009
Jaworski A, Osteryoung JG, Donten M, Stojek Z (1999) The strength of acids in alcohols as determined by steady-state voltammetry. Anal Chem 71:3853–3861. https://doi.org/10.1021/ac9814593
de Souza FC, da Rocha JC, Vieira E et al (2009) Development of an analytical methodology for quantification of strong acid in diesel oil. Electroanalysis 21:2277–2280. https://doi.org/10.1002/elan.200900143
Baldo MA, Oliveri P, Simonetti R, Daniele S (2016) A novel electroanalytical approach based on the use of a room temperature ionic liquid for the determination of olive oil acidity. Talanta 161:881–887. https://doi.org/10.1016/j.talanta.2016.09.045
Silvester DS, He W, Aldous L et al (2008) Electrochemical reduction of benzoic acid and substituted benzoic acids in some room temperature ionic liquids. J Phys Chem C 112:12966–12973. https://doi.org/10.1021/jp802996q
He W, Silvester DS, Streeter I et al (2009) Measuring the solubility of benzoic acid in room temperature ionic liquids using chronoamperometric techniques. J Phys Org Chem 22:69–76. https://doi.org/10.1002/poc.1428
Meng Y, Norman S, Hardacre C, Compton RG (2013) The electroreduction of benzoic acid: voltammetric observation of adsorbed hydrogen at a platinum microelectrode in room temperature ionic liquids. Phys Chem Chem Phys 15:2031–2036. https://doi.org/10.1039/C2CP43580B
Ledezma-Yanez I, Díaz-Morales O, Figueiredo MC, Koper MTM (2015) Hydrogen oxidation and hydrogen evolution on a platinum electrode in acetonitrile. ChemElectroChem 2:1612–1622. https://doi.org/10.1002/celc.201500341
Bentley CL, Bond AM, Hollenkamp AF et al (2015) Electrochemical proton reduction and equilibrium acidity (pKa) in aprotic ionic liquids: protonated amines and sulfonamide acids. J Phys Chem C 119:21828–21839. https://doi.org/10.1021/acs.jpcc.5b05723
Bentley CL, Bond AM, Hollenkamp AF et al (2015) Electrochemical proton reduction and equilibrium acidity (pKa) in aprotic ionic liquids: phenols, carboxylic acids, and sulfonic acids. J Phys Chem C 119:21840–21851. https://doi.org/10.1021/acs.jpcc.5b05724
Bentley CL, Bond AM, Zhang J (2018) Voltammetric perspectives on the acidity scale and H+/H2 process in ionic liquid media. Annu Rev Anal Chem 11:397–419. https://doi.org/10.1146/annurev-anchem-061417-010022
Daniele S, Ugo P, Mazzocchin G-A, Bontempelli G (1985) Acid-base equilibria in organic solvents: part 1. Evaluation of solvent basicity by cyclic voltammetry. Anal Chim Acta 173:141–148. https://doi.org/10.1016/S0003-2670(00)84951-X
Kurek SS, Laskowska BJ, Stokłosa A (2006) Cathodic reduction of acids in dimethylformamide on platinum. Electrochim Acta 51:2306–2314. https://doi.org/10.1016/j.electacta.2005.04.085
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Baldo, M.A., Fabris, S., Stortini, A.M. et al. Protolysis studies and quantification of acids and bases in aqueous solutions by microelectrode voltammetry. J Solid State Electrochem 28, 1049–1068 (2024). https://doi.org/10.1007/s10008-023-05675-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-023-05675-8