
Abstract
A facile strategy was developed to prepare interlayer-expanded MoS2/graphene composites through a one-step hydrothermal reaction method. MoS2 nanosheets with several-layer thickness were observed to uniformly grow on the surface of graphene sheets. And the interlayer spacing of MoS2 in the composites was determined to expand to 0.95 nm by ammonium ions intercalation. The MoS2/graphene composites show excellent lithium storage performance as anode materials for Li-ion batteries. Through gathering advantages including expanded interlayers, several-layer thickness, and composited graphene, the composites exhibit reversible capacity of 1030.6 mAh g−1 at the current density of 100 mA g−1 and still retain a high specific capacity of 725.7 mAh g−1 at a higher current density of 1000 mA g−1 after 50 cycles.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The growing concerns on energy shortage and ecological problems from burning fossil fuels have greatly promoted the exploration on clean and renewable energy sources. Lithium-ion batteries (LIBs) with high energy and power density and long cycle life, as one important power sources for portable electric devices, have been demonstrated to be a critical part of the energy sources in the future [1, 2]. For commercial applications, graphite is widely utilized as the anode material for LIBs because of its flat potential profile, high columbic efficiency, and good cycling performance. However, the energy density is limited by its low theoretical specific capacity (372 mAh g−1), and a low power density is always obtained resulting from slow Li+ diffusion in the graphite structure [3]. Thus, the realization of alternative anode materials with higher capacity has recently attracted much effort to establish the next generation of commercial LIBs with better performance.
Among the numerous developed candidates, MoS2 has exhibited especially outstanding lithium storage performance due to its unique structure and property [4, 5]. As a typical layered transition metal sulfide, MoS2 is constructed by three atom layers (S-Mo-S) stacked together through van der Waals interactions which can be exfoliated into nanosheets with fewer layers during the first Li insertion [6]. Such transformation can facilitate the following Li diffusion and accommodate Li integrations with significantly less volumetric expansion on lithiation/delithiation cycling process, therefore offering the advantages to result in desirable rate capacity and cycling stability. And compared with the intercalation mechanism-based materials (e.g., graphite), MoS2 can deliver higher energy density according to its conversion reaction mechanism in LIBs [7]. Despite the advantages of MoS2 as anode material for LIBs, the performance is still unsatisfactory for large-scale practical applications. To further improve the performance of MoS2-based LIBs, various strategies have been investigated, mainly including enlarging the interlayer distance of MoS2 [8,9,10], constructing hierarchical nanostructures [11] and MoS2/carbon composites [12,13,14,15]. Increasing the space between MoS2 interlayers can lower the Li+ insertion and diffusion barriers and accommodate more Li+. Constructing hierarchical nanostructures of MoS2 is able to shorten the distance of Li+ diffusion and as well increase the contact area between electrode and electrolyte. As for MoS2/carbon composites, the composition of high conductive carbonaceous materials is always designed to remedy the relatively poor conductivity of MoS2 semiconductor. Especially, graphene has always been selected as promising matrices due to its large surface area, high electric conductivity, and good chemical stability [16,17,18,19,20]. Obviously, any above aspect of engineering the structure of MoS2 can make a positive contribution to improve the lithium storage performance, while designing versatile structures that can gather all the advantages is highly desired and efficient.
Herein, we construct interlayer-expanded MoS2/graphene composites via a one-step hydrothermal reaction method. The results demonstrate that MoS2 nanosheets are just several-layer thick and uniformly grown on the surface of graphene. And the interlayer of MoS2 in the composites is expanded to 0.95 nm by intercalated ammonium ions. Although ammoniated MoS2 through hydrothermal route was recently reported by usually selecting thioacetamide (CH3CSNH2)/thiourea (NH2CSNH2) as S precursor, l-cysteine is rarely utilized to synthesize ammoniated MoS2. With l-cysteine in this work, we successfully expand the MoS2 interlayers through intercalating ammonium ions during the MoS2 growing on graphene. Interestingly, our ammoniated MoS2 exhibit much better structure stability compared with previous results which is benefit for their application as anode in LIBs. Based on gathering the advantages of expanded interlayers, several-layer thickness, and composited graphene, the MoS2/graphene composites exhibit higher capacity of 1030.6 mAh g−1 than that of bare MoS2 at the current density of 100 mA g−1 after 50 cycles. More significantly, a high capacity of 725.7 mAh g−1 is achieved at a higher current density of 1000 mA g−1.
Experiment section
Synthesis of MoS2/graphene composites
Graphene oxide (GO) nanosheets were fabricated via the oxidation of graphite using a modified Hummers’ method [21]. To prepare the MoS2/graphene composites, 0.18 g of Na2MoO4· 2H2O and 0.45 g l-cysteine were dissolved in 40 mL as-prepared GO suspension (0.1 g/L, in deionized water). The mixture was kept stirring for 1 h and then transferred into a 50-mL Teflon-lined stainless steel autoclave and heated at 200 °C for 40 h. Controlled experiments were conducted under the same conditions except for different reaction temperatures from 180 to 240 °C. After cooling naturally, the products were collected by centrifugation, washed with deionized water and ethanol, and dried in a vacuum oven at 60 °C for overnight. Bare MoS2 were prepared under the same conditions without adding GO at 200 °C for 40 h.
Materials characterization
The X-ray powder diffraction (XRD) patterns were obtained by using a Rigaku D/max 2500 X-ray diffractometer with Cu Kαradiation (λ = 1.5418 Å) at 40 kV. The morphologies of the samples were observed through scanning electron microscopy (SEM, Verios 460L field emission instrument) and transmission electron microscopy (TEM, Talos F200X) measurements. Thermogravimetric analyses (TGA) and differential scanning calorimeter (DSC) measurement were conducted on TG 209 F3 and DSC 214, respectively. Raman spectra were achieved by Renishaw in Via spectrometer (514.5-nm laser). Fourier transform infrared spectroscopy (FTIR) analyses were performed on Frontier Mid-IR FTIR. The chemical states of the compounds were investigated through XPS by using Axis Ultra DLD, Kratos Analytical.
Electrochemical measurements
Electrochemical measurements were conducted on two-electrode coin cells (Type 2025). The MoS2/graphene, acetylene black, and poly (vinylidene fluoride) binder were mixed at a weight ratio of 80:10:10 and dispersed in N-methylpyrrolidone. The mixture was kept stirring overnight to form a homogenous slurry. Then, the slurry was firstly coated on copper foil and dried in a vacuum oven at 80 °C for 2 h and then heated up to 120 °C overnight. The electrode foil was punched into 12-mm-diameter discs prior to coin-cell assembly. The average weight of the active materials on the discs is ~ 0.9 mg. In the test cells, lithium metal was utilized as the counter and reference electrode, Celgard 2300 was chosen as the separator. The 1 M LiPF6 electrolyte was prepared by dissolving LiPF6 in a mixture of ethylene carbonate, ethylene methyl carbonate and dimethyl carbonate by volume of 1:1:1. The cells were assembled in a glove box filled with high-purity argon. The galvanostatic charge and discharge tests were performed with a battery tester LAND-CT2001A in the voltage range of 0.001–3.0 V.
Results and discussion
Structural verification
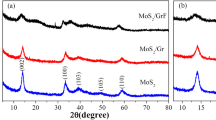
The X-ray diffraction (XRD) patterns of the samples, including a series of MoS2/graphene composites, synthesized bare MoS2 (S-MoS2) and commercially available MoS2 (pristine MoS2) are exhibited in Fig. 1. The results in Fig. 1a show that the crystalline structure of MoS2 in MoS2/graphene composites prepared at 200 °C is consistent with that of synthesized MoS2 without GO. Specifically, in the inset in Fig. 1a, the diffraction peaks corresponding to the interlayer spacing of MoS2 in MoS2/graphene composites and S-MoS2 both obviously shift to a lower angle (2θ = 9.3°) compared with that of pristine MoS2 (2θ = 14.4°). And the first two reflecting peaks of MoS2 have a diploid relationship (2θ = 9.3° and 18.1°), demonstrating the existence of a multiple MoS2 molecular layers stacked periodically. According to the Bragg’s equation, nλ = 2dsinθ (n, λ, d and θ represent a positive integer, wavelength of incident X-ray, interplanar distance of lattice, and diffraction angle, respectively), the shift indicates that the interlayers of MoS2 are expanded and the spacing increases to 0.95 nm (0.615 nm [22] for pristine MoS2). The increased distance of 0.335 nm matches well with the size of NH4+ whose hydrogen-bonding diameter is about 0.35 nm [23]. Notably, the expanded lamellar structure can be constructed under a wide range of reaction temperature from 180 to 240 °C as shown in Fig. 1b. The expanded interlayers can be obtained even at high reaction temperature, indicating the good thermal stability of the product. The interlayer expansion with good stability is expected to enhance the accommodation of Li ions in the interlayer gaps of MoS2/graphene composites without significant volume variation during charge/discharge cycles, therefore resulting in large specific capacity and high cycle stability.

XRD patterns of a MoS2/graphene composites synthesized at 200 °C, S-MoS2 and pristine MoS2 and b a series of MoS2/graphene composites obtained under different reaction temperatures
Figure 2a compares the FTIR results of interlayer-expanded MoS2/graphene composites, annealed composites under 800 °C and pristine MoS2. All the spectra show two apparent adsorption peaks located at 3440 and 1636 cm−1 which can be assigned to the stretching and bending vibrations of adsorbed water, respectively [24]. After the composites were annealed at 800 °C in N2 atmosphere, the adsorption peaks are almost consistent with that of pristine MoS2, illustrating a structure transformation to stable 2H-MoS2 under thermal treatment. The region from 1000 to 1200 cm−1 mainly corresponds to S-O/S=O and Mo = O bands due to partial surface oxidation that has also been observed previously [25]. And the peak at 622 cm−1 should be due to the Mo-S vibration [24]. By comparison, an obvious adsorption peak at 1400 cm−1 is only present in the interlayer-expanded MoS2/graphene composites but absent in others. The adsorption is assigned to the asymmetric bending vibration of NH4+ inserted in the interlayers of MoS2, while the invisible vibration of N-H at about 3200 cm−1 is due to the coverage by O-H vibration [26]. The Raman spectra of GO and MoS2/graphene composites in Fig. 2b exhibit two obvious bands around 1350 and 1588 cm−1, assigning to the breathing mode of A1g symmetry (D band) and first-order scattering of E2g phonons (G band) of carbon materials, respectively. The D band is related to structural defects or partially disordered arrangements in graphitic materials and the G band is associated with the graphitic carbon. The intensity ratio of D band to G band (Id/Ig) is increased from 1.04 for GO to 1.27 for MoS2/graphene composites, indicating an increase in the number of smaller conjugated domains which is usually observed in the Raman spectra of reduced graphene oxide [27].

a FTIR spectra of MoS2/graphene composites, annealed composites and pristine MoS2. b Raman spectra of MoS2/graphene composites and GO
TGA/DSC analyses were conducted to further characterize the structure of MoS2/graphene composites. From the TGA result in Fig. 3a, the MoS2 content in the composites is calculated to be 74.9 wt% assuming all the graphene is combusted by O2 in air and the MoS2 is completely converted to MoO3 [28]. Under N2 atmosphere in Fig. 3b, the MoS2/graphene composites exhibit two mass loss steps. The first weight loss of about 7% centered at 70 °C is mostly resulted from the release of inserted water/NH3 [29]. And the second loss of 14.5% centered at 300 °C associated with an endothermic peak is resulted from the release of intercalated NH4+. Annealed composites at 300 °C give a similar XRD pattern with that of pristine MoS2 (Fig. S1), indicating a structure conversion to thermodynamically stable 2H-MoS2 following the deintercalation of NH4+. Importantly, the structure conversion happens at a higher temperature than the ones of previously reported ammoniated MoS2 (usually around 240 °C [29, 30]), demonstrating a good thermal stability of the present composites.

a TGA profile of MoS2/graphene composites conducted in air. b TGA/DSC profiles of MoS2/graphene composites conducted in N2
XPS spectra are further analyzed to investigate the chemical composition of the ammoniated MoS2/graphene composites. Figures S2A and 4A compare the C 1s XPS peak-fitting results of graphene oxide and MoS2/graphene composites. As exhibited in Fig. S2A, four resolved peaks located at 286.9, 287.4, 288.7, and 284.8 eV refer to oxygenated functional groups of C–O, C=O, O–C=O, and sp2-hydridized C–C bonds, respectively, indicating a considerable degree of oxidation in the graphene oxide [31]. By comparison, in Fig. 4a, the C 1s spectrum of MoS2/graphene composites shows much weaker peaks of corresponding oxygenated functional groups. The obviously decreased peaks illustrate that the graphene oxide have been effectively reduced during the composition process with MoS2 nanosheets under high reaction temperature and pressure [32]. The reduced graphene oxide is expected to increase the conductivity of MoS2 and thus improve the performance of the composites as the anode material in LIBs. In the Mo 3d spectrum of MoS2/graphene composites (Fig. 4b), five peaks are attributed to Mo6+, Mo 3d3/2, Mo 3d5/2, and S 2s peaks [33]. The Mo6+ peaks located at 236.6 and 233.5 eV referring to Mo-O can be ascribed to partial oxidation of surface Mo atoms. The two main peaks of Mo 3d3/2 (232.6 eV) and Mo 3d5/2 (229.1 eV) are characteristic of MoS2 and 226.7 eV corresponds to S 2s of MoS2. Figure 4c shows the sulfur species of MoS2. The doublet peaks at 163.5 and 162.3 eV refer to the S 2p1/2 and S 2p3/2 of MoS2. The S6+ peak (169.4 eV) is possibly due to the oxidation of S2− in the atmosphere [34]. What’s more, the binding energy located at 402 eV in N 1s spectrum obviously demonstrates the existence of NH4+ ions [29], further indicating the construction of ammoniated MoS2 composites. The calculated weight percentage of N element of 9.83% by XPS in Table S1 gives a content of 12.64 wt% of inserted NH4+ which is consistent with the result of TGA. After annealed at 800 °C, completed remove of inserted ammonium ions is indicated by the absent corresponding signal of N 1s (Fig. S2B).

XPS spectra of the ammoniated MoS2/graphene composites. a C 1s spectrum. b Mo 3d spectrum. c S 2p spectrum. d N 1s spectrum
Morphology of the MoS2/graphene composites
The general morphology of the MoS2/graphene composites is exhibited in Fig. 5. As shown in the SEM image in Fig. 5a, a composite structure of MoS2 and graphene is achieved instead of separately grown structures. The TEM image in Fig. 5b further demonstrates that MoS2 nanosheets are uniformly grown on the surface of graphene sheets. However, large spheres with a size of ~ 700 nm assembled by MoS2 nanosheets are obtained without adding GO in the synthesis process (Fig. S3), indicating the effectiveness of the present method to disperse MoS2 nanosheets on graphene. Figure 5c depicts a high-resolution TEM image of the MoS2/graphene nanocomposites which confirms an average interlayer distance of 0.93 nm in the MoS2 nanosheets, in consistent with the value calculated from XRD data. The elemental mapping image in Fig. 5d further demonstrates the homogeneous distribution of Mo, S, and C in the composites.

SEM (a), TEM (b), HRTEM (c), and element mapping (d) images of MoS2/graphene composites
Lithium storage performance of the MoS2/graphene composites
The lithium storage properties of the MoS2/graphene composites are first characterized by discharge/charge voltage profiles of 1st, 2nd, 10th, 20th, and 50th cycles at a current density of 100 mA g−1 (Fig. 6a). As shown in Fig. 6a, the MoS2/graphene electrode delivers high first-cycle discharge and charge capacities of 2037 and 1690 mAh g−1 respectively, corresponding to a high initial Coulombic efficiency (CE) of 83.0%. The irreversible loss derives from the formation of solid-electrolyte interface films and the electrolyte decomposition [35]. In the second cycle, a discharge/charge capacity of 1624/1735 mAh g−1 gives a higher CE of 93.6%. Moreover, the discharge capacity remains at 1030.6 mAh g−1 after 50 cycles, which is much higher than 50.1 mAh g−1 of pristine MoS2 (Fig. 6b). Importantly, the layered structure of MoS2 in the composites can be retained after charge/discharge cycles, which is demonstrated by the SEM image of anode electrode from cycled coin-cell (Fig. S4A). The structure stability of the MoS2 composites is mostly attributed to the expanded interlayers with good accommodation of volume variation and the contribution of composited graphene. Without graphene, the morphology of bare MoS2 shows no obvious layered structure after cycles (Fig. S4B). Remarkably, at a higher current density of 1000 mA g−1, the MoS2/graphene composites still exhibit excellent cycling performance (a discharge capacity of 725.7 mAh g−1 after 50 cycles). As comparison, the physical mixture of MoS2 and graphene (about 75 wt% of MoS2, equal to that of MoS2/graphene composites) only retains 421.8 mAh g−1, further indicating the effectiveness of the in situ growth of MoS2 on the surface of graphene to improve the performance of the composites.

a Discharge/charge curves of 1st, 2nd, 10th, 20th, and 50th cycles for MoS2/graphene composites. b Cycling performance of MoS2/graphene composites, S-MoS2 and the mixture of MoS2 and graphene. c Rate capability of the MoS2/graphene at different current densities. d Nyquist plots of MoS2/graphene composites and S-MoS2 electrodes
Figure 6c exhibits the rate capacity of MoS2/graphene electrode at different current densities ranging from 100 to 2000 mA g−1. The composites deliver the discharge capacities of 1066.4, 1041.3, 985.2, 890.5, and 745.2 mAh g−1 at current densities of 100, 200, 500, 1000, and 2000 mA g−1, respectively. When the current density is returned back to 100 mA g−1, the capacity rapidly recovers to 1236.7 mAh g−1, probably resulting from the growth of a gel-like polymeric layer and activation of electrode materials [36]. Electrochemical impedance spectroscopy (EIS) of the MoS2/graphene composites and S-MoS2 is performed to further understand the enhanced electrochemical performance. Figure 6d compares the resistances of the MoS2/graphene composites and S-MoS2. The result shows that the resistance of the MoS2/graphene composites is much smaller than that of S-MoS2, demonstrating a better electronic transportation after compositing graphene.
The improved electrochemical performance of the MoS2/graphene composites might arise from a synergistic effect between MoS2 and graphene: (i) MoS2 nanosheets uniformly distribute on the surface of graphene, the graphene can restrain the aggregation of MoS2 nanosheets during lithiation/delithiation process. (ii) Our in situ growth strategy ensures a good contact between the MoS2 and graphene, thus enhancing the electronic transportation in the composites. (iii) The expansion of MoS2 interlayer weakens the van der Waals interaction between the layers, thus promoting the Li+ intercalation and accommodate the corresponding volume variation. Thanks to the advantages of our designed structure, the lithium storage properties of the MoS2/graphene composites are more desirable compared with the S-MoS2 and the physical mixture of MoS2 and graphene. Specially, as shown in Table S2, the MoS2/graphene composites in this work show comparable cycling performance with reported MoS2/carbon composites at the current density of 100 mA g−1 and even better stability under larger current density (1000 mA g−1 in our case).
In summary, ammoniated MoS2/graphene composites with expanded interlayers have been successfully constructed via a one-step hydrothermal method. The MoS2/graphene composites exhibit excellent lithium storage performance with high reversible capacity and good cycling stability even at high current density. The simple strategy, effective composite structure and outstanding lithium storage properties are believed to inspire the establishment of next generation of LIBs with promising performance.
References
Nishi Y (2001) The development of lithium ion secondary batteries. Chem Rec 1(5):406–413
Kraytsberg A, Ein-Eli Y (2017) A critical review-promises and barriers of conversion electrodes for Li-ion batteries. J Solid State Electrochem 21(7):1907–1923
Li H, Wang Z, Chen L, Huang X (2009) Research on advanced materials for Li-ion batteries. Adv Mater 21(45):4593–4607
Xu X, Liu W, Kim Y, Cho J (2014) Nanostructured transition metal sulfides for lithium ion batteries: progress and challenges. Nano Today 9(5):604–630
Sen UK, Mitra S (2014) Improved electrode fabrication method to enhance performance and stability of MoS2-based lithium-ion battery anode. J Solid State Electrochem 18(10):2701–2708
Zeng Z, Yin Z, Huang X, Li H, He Q, Lu G, Boey F, Zhang H (2011) Single-layer semiconducting nanosheets: high-yield preparation and device fabrication. Angew Chem Int Ed 50(47):11093–11097
Du G, Guo Z, Wang S, Zeng R, Chen Z, Liu H (2010) Superior stability and high capacity of restacked molybdenum disulfide as anode material for lithium ion batteries. Chem Commun 46(7):1106–1108
Rasamani KD, Alimohammadi F, Sun Y (2017) Interlayer-expanded MoS2. Mater Today 20(2):83–91
Guo J, Zhu H, Sun Y, Tang L, Zhang X (2016) Boosting the lithium storage performance of MoS2 with graphene quantum dots. J Mater Chem A 4(13):4783–4789
Xiao J, Choi D, Cosimbescu L, Koech P, Liu J, Lemmon JP (2010) Exfoliated MoS2 nanocomposite as an anode material for lithium ion batteries. Chem Mater 22(16):4522–4524
Wang P p, Sun H, Ji Y, Li W, Wang X (2014) Three-dimensional assembly of single-layered MoS2. Adv Mater 26:964–969
Gao M-R, Xu Y-F, Jiang J, Yu S-H (2013) Nanostructured metal chalcogenides: synthesis, modification, and applications in energy conversion and storage devices. Chem Soc Rev 42(7):2986–3017
Guo Z-Y, Zhong Y, Liu Y, Mao C-M, Li G-C (2017) MoS2 nanosheet arrays supported on hierarchical porous carbon with enhanced lithium storage properties. Chin Chem Lett 28(4):743–747
Mao C, Zhong Y, Shang H, Li C, Guo Z, Li G (2016) Carbon encapsulated nanosheet-assembled MoS2 nanospheres with highly reversible lithium storage. Chem Eng J 304:511–517
Zhong Y, Zhuang Q, Mao C, Xu Z, Guo Z, Li G (2018) Vapor phase sulfurization synthesis of interlayer-expanded MoS2@ C hollow nanospheres as a robust anode material for lithium-ion batteries. J Alloys Compd 745:8–15
Jiang L, Lin B, Li X, Song X, Xia H, Li L, Zeng H (2016) Monolayer MoS2–graphene hybrid aerogels with controllable porosity for lithium-ion batteries with high reversible capacity. ACS Appl Mater Interfaces 8(4):2680–2687
Chang K, Chen W (2011) L-cysteine-assisted synthesis of layered MoS2/graphene composites with excellent electrochemical performances for lithium ion batteries. ACS Nano 5(6):4720–4728
Gong Y, Yang S, Zhan L, Ma L, Vajtai R, Ajayan PM (2014) A bottom-up approach to build 3D architectures from nanosheets for superior lithium storage. Adv Funct Mater 24(1):125–130
Jing Y, Ortiz-Quiles EO, Cabrera CR, Chen Z, Zhou Z (2014) Layer-by-layer hybrids of MoS2 and reduced graphene oxide for lithium ion batteries. Electrochim Acta 147:392–400
Cao X, Shi Y, Shi W, Rui X, Yan Q, Kong J, Zhang H (2013) Preparation of MoS2-coated three-dimensional graphene networks for high-performance anode material in lithium-ion batteries. Small 9(20):3433–3438
Hummers WS Jr, Offeman RE (1958) Preparation of graphitic oxide. J Am Chem Soc 80(6):1339–1339
Dungey KE, Curtis MD, Penner-Hahn JE (1998) Structural characterization and thermal stability of MoS2 intercalation compounds. Chem Mater 10(8):2152–2161
Ong EW, Eckert J, Dotson LA, Glaunsinger WS (1994) Nature of guest species within alkaline earth-ammonia intercalates of titanium disulfide. Chem Mater 6(11):1946–1954
Huang K-J, Wang L, Zhang J-Z, Wang L-L, Mo Y-P (2014) One-step preparation of layered molybdenum disulfide/multi-walled carbon nanotube composites for enhanced performance supercapacitor. Energy 67:234–240
Maugé F, Lamotte J, Nesterenko N, Manoilova O, Tsyganenko A (2001) FT-IR study of surface properties of unsupported MoS2. Catal Today 70(1-3):271–284
Liu S, Zhang X, Shao H, Xu J, Chen F, Feng Y (2012) Preparation of MoS2 nanofibers by electrospinning. Mater Lett 73:223–225
Wang H, Robinson JT, Li X, Dai H (2009) Solvothermal reduction of chemically exfoliated graphene sheets. J Am Chem Soc 131(29):9910–9911
Zhao C, Wang X, Kong J, Ang JM, Lee PS, Liu Z, Lu X (2016) Self-assembly-induced alternately stacked single-layer MoS2 and N-doped graphene: a novel van der Waals heterostructure for lithium-ion batteries. ACS Appl Mater Interfaces 8(3):2372–2379
Anto JA, Nethravathi C, Rajamathi M (2014) Two-dimensional nanosheets and layered hybrids of MoS2 and WS2 through exfoliation of ammoniated MS2 (M= Mo, W). J Phys Chem C 118(2):1386–1396
Wu Z, Tang C, Zhou P, Liu Z, Xu Y, Wang D, Fang B (2015) Enhanced hydrogen evolution catalysis from osmotically swollen ammoniated MoS2. J Mater Chem A 3(24):13050–13056
Chen J, Sheng K, Luo P, Li C, Shi G (2012) Graphene hydrogels deposited in nickel foams for high-rate electrochemical capacitors. Adv Mater 24(33):4569–4573
Li H, Yu K, Fu H, Guo B, Lei X, Zhu Z (2015) MoS2/graphene hybrid nanoflowers with enhanced electrochemical performances as anode for lithium-ion batteries. J Phys Chem C 119(14):7959–7968
Chao Y, Jalili R, Ge Y, Wang C, Zheng T, Shu K, Wallace GG (2017) Self-assembly of flexible free-standing 3D porous MoS2-reduced graphene oxide structure for high-performance lithium-ion batteries. Adv Funct Mater 27(22). https://doi.org/10.1002/adfm.201700234
Wang S, Tu J, Yuan Y, Ma R, Jiao S (2016) Sodium modified molybdenum sulfide via molten salt electrolysis as an anode material for high performance sodium-ion batteries. Phys Chem Chem Phys 18(4):3204–3213
Zhen M, Zhen X, Liu L (2017) Mesoporous nanoplate TiO2/reduced graphene oxide composites with enhanced lithium storage properties. Mater Lett 193:150–153
Xu X, Fan Z, Yu X, Ding S, Yu D, Lou XWD (2014) A nanosheets-on-channel architecture constructed from MoS2 and CMK-3 for high-capacity and long-cycle-life lithium storage. Adv Energy Mater 4(17). https://doi.org/10.1002/aenm.201400902
Funding
This work was supported by the National Natural Science Foundation of China (NSFC 21571170, 21501168, 21701124, and 51702236) and Postdoctoral Science Foundation of China (2017M611171).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
ESM 1
(DOCX 889 kb)
Rights and permissions
About this article

Cite this article
Wang, Y., Zhen, M., Liu, H. et al. Interlayer-expanded MoS2/graphene composites as anode materials for high-performance lithium-ion batteries. J Solid State Electrochem 22, 3069–3076 (2018). https://doi.org/10.1007/s10008-018-4018-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-018-4018-8