
Abstract
The effect of sodium dodecyl sulfate and carbon particles/nanotubes on the electropolymerization of aniline from oxalic acid solution onto a graphite electrode was investigated. The morphology and chemical structure of the as-synthesized polyaniline films were studied by means of SEM, XPS, NEXAFS, FTIR, and Raman spectroscopy. The electrochemical characteristics of the films were also analyzed in sulfuric acid solution. It has been shown that in the presence of sodium dodecyl sulfate, the polymerization rate increases significantly. In the synthesized polyaniline films, all imine groups and most of amine groups are protonated, with dodecyl sulfate ions being intercalated in the polymer. In the presence of sodium dodecyl sulfate, plate-like polyaniline forms large agglomerates with an extended surface and high electrochemical activity. It has been shown that the electrodeposition carried out in the presence of sodium dodecyl sulfate and suspended activated graphite particles or carbon nanotubes favors the formation of composite coatings with high specific capacitance.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polyaniline (PANI) is a well-studied conductive polymer used in electrochemical devices (batteries, fuel cells, electrochemical capacitors), membranes, sensors, electrocatalysts, electromagnetic shields, anti-corrosion and anti-electrostatic coatings, and electrochromic devices [1,2,3,4,5,6,7]. As functional coatings, PANI can be produced by both chemical oxidation of aniline and its electrochemical oxidation on various conductive substrates [3, 4]. To date, it has been found that the physical and chemical properties of PANI (processability, mechanical properties, electrical conductivity, thermal stability, and redox behavior) can be significantly improved by doping with alkyl sulfates, including widely used sodium dodecyl sulfate (SDS) [8,9,10,11,12,13,14]. For electrodeposited PANI films, SDS additives lead to the intercalation of the surfactant molecules into the polymer, changing the morphology and electrochemical characteristics of the films [15,16,17]. As an example, such doping improves the characteristics of films for their use as sensors [18, 19].
The effect of SDS additives on the electrochemical synthesis of polyaniline was earlier studied for the strong acid (HCl, H2SO4) solutions [15,16,17,18]. The published data concern the morphology and electrochemical characteristics of doped films, whereas their chemical structure has not been comprehensively considered. There is no data on the effect of SDS additives on the electrochemical synthesis of PANI and its chemical structure when depositing from the solutions of moderate acids, such as oxalic acid. Oxalic acid is often used when depositing protective films of conductive polymers onto the surface of iron and steel [6, 20].
Besides, SDS is a very good dispersing agent successfully used for the stabilization of suspensions of carbon nanotubes and nanoparticles [21, 22]. As a result, an addition of SDS provides electrochemical deposition of PANI in suspensions of carbon particles and thus fabrication of composite coatings. Composite materials based on PANI with carbon particles or carbon nanotubes (CNTs) are of great potential for practical application as electrochemical capacitors, sensors, and materials for fuel cells [4, 23].
In this paper, the effect of SDS on the electrodeposition of PANI from oxalic acid solutions onto a graphite substrate was investigated. The chemical structure of the electrodeposited PANI films was studied by means of XPS, NEXAFS, Raman, and FTIR spectroscopies. Electrochemical characteristics of the as-deposited films were also measured. In addition, the electrodeposition was applied to synthesize composite films of PANI from suspensions of carbon particles (mechanically activated graphite) or CNTs stabilized with SDS additives. The chemical structure and electrochemical characteristics of the composite films were also thoroughly investigated.
Experimental
The PANI films were electrochemically deposited on high pure polycrystalline graphite, with the impurity concentration not exceeding 5 10−3 wt%. Prior to the deposition, the substrates of 0.25 cm2 area had been thoroughly treated with SiC-based polishing paper (1500 grit), degreased with ethanol and then washed with distilled water. The polarization was performed in naturally aerated and moderately stirred solution of 0.3 M oxalic acid containing 0.1 M aniline (pH = 1.0).
The films were deposited and then studied at Potentiostat Pi-50-Pro (Elins) with a glass cell. The auxiliary and reference electrodes were Pt and Ag/AgCl, respectively, located in separate compartments. Hereinafter, the potentials are given relative to the Ag/AgCl reference electrode. The solution volume for the film synthesis was 80 ml. The samples were mounted vertically in the cell. The stirring of the solution was carried out with a magnetic stirrer of a cylindrical shape with the diameter of 7 mm and length of 33 mm. The rotation speed was 350 rpm.
The films were deposited using potentiodynamic polarization (50 cycles) in the range of 300–1200 mV at a rate of 25 mV s−1. The same scan rate and potential range had been successfully used earlier to grow polyaniline films on iron substrates [20]. The potential range chosen for the synthesis is characterized by high value of positive potential limit. The polarization in the solutions of strong acids (HCl, H2SO4) is usually performed up to 750–800 mV vs. Ag/AgCl to avoid overoxidation and then destruction of the polymer [24,25,26,27]. However, in the case of stirred solution of oxalic acid used as an electrolyte, similar conditions did not lead to the precipitation of polyaniline film on the graphite electrode surface. This may be explained by stirring the solution, which significantly reduces the rate of polyaniline precipitation due to the removal of intermediate oxidation products and oligomers from the surface [27]. The acidity of the oxalic acid solution was probably not sufficient for the polymerization in stirred solution, since the polymerization rate decreases on going from strong acid (pH = 0) to less acidic (pH = 1–2) solutions [28].
SDS additive was introduced as a concentrated 1 M aqueous solution (5 ml) after 1000 s of polarization (14th cycle). Carbon particles or CNTs in amount of 100 mg were added into the solution if it was the case. So, the concentration of SDS in the solution prepared for depositing PANI was 0.059 M. The first stage of deposition was carried out without SDS additives for better adhesion of the films to the substrates. After synthesis, the samples were immediately rinsed with distilled water to remove electrolyte from the surface and dried over KOH in an argon-filled box. Prior to measurements, the samples were stored and transported in an argon-filled box.
To fabricate composite coatings, multiwall CNTs and mechanically activated graphite were incorporated into the films. CNTs were produced by the chemical vapor deposition (CVD) technique described in detail elsewhere [30]. Carbon particles were produced by mechanical activation of polycrystalline graphite (with the impurity concentration not exceeding 5 10−3 wt%) in a planetary ball mill Fritsch P7. The milling time was 8 h. Vials (45 cm3) and balls (16 pcs, diameter 12 mm) of the mill are made from the hardened steel containing 1.5 wt% of chromium and 1.0 wt% of carbon. Graphite in amount of 3 g was put into a vial, the rest space of which was then filled with heptane (first case) or 20 wt% aqueous solution of ammonium persulfate (second case). The temperature of the vial outer walls was not higher than 60 °C due to the forced air-cooling. After milling, the graphite particles were separated by decantation and washed with the appropriate solvent (heptane or distilled water). Because of noticeable oxidation of the vial and the balls in aqueous medium, some parts of the powder was thoroughly rinsed with acid solutions (5 wt% HCl, HNO3) and then washed with distilled water.
The films deposited without and with SDS additives will be designated as Pani and Pani/SDS, respectively. Composite films will be denoted depending on the type of the incorporated particles as Pani/SDS_CNT (carbon nanotubes), Pani/SDS_G H (graphite particles mechanoactivated in heptane), Pani/SDS_G A (graphite particles mechanoactivated in aqueous solution of ammonium persulfate), and Pani/SDS_G A * (graphite particles mechanoactivated in aqueous solution of ammonium persulfate then additionally washed in acidic solutions).
The surface morphology and topography of the deposited films were examined by scanning electron microscopy with a VEGA 3 LMH (TESCAN) microscope. Raman spectra were acquired with a Horiba LabRam HR800 spectrometer using a He-Ne laser operating at a wave length of 632.8 nm and input power not exceeding 10 mW at the sample surface. The spectrometer was equipped with an optical microscope (×100 objective lens) allowing one to focus the laser beam on different sites of the sample surface. The beam diameter in the analysis was about 5 μm. Neutral filters were used to reduce laser beam power and avoid degradation of the films.
The films for the FTIR analysis were thoroughly separated from the substrates. Most of the films were relatively thick and loose and therefore easily separated from the graphite substrate surface with a scalpel. The as-separated film was thoroughly mixed with KBr and then pressed into a pellet. FTIR spectra were measured in the transmission mode with an Excalibur Varian 3100 FT-IR spectrometer. The spectra were collected with a resolution of 1 cm−1. For the Pani sample, an amount of the polymer was too little to measure the spectrum. In this case, a comparison is made with the film deposited under the same conditions on iron substrate. Before deposition, the substrate was thoroughly polished and then potentiodynamically treated to form a passivating oxalate layer as proposed in [20]. The spectrum was measured with an accessory for measuring the spectra in specular reflection mode with an incident angle 10°. The measurement range was up to 2000 cm−1 in this case.
The NEXAFS spectra were acquired at the Russian-German beamline (RGBL) at the BESSY II, Helmholtz-Zentrum Berlin (HZB), Berlin GmbH. The beamline equipped with a photoemission end-station allows one to carry out measurements of the spectra in the photon energy range from 40 to 1500 eV in ultrahigh vacuum (~ 5 × 10−8 Pa). The NEXAFS spectra were obtained in a total electron yield (TEY) mode by measuring the drain current from the sample changing the incident photon energy. The detailed description of the beamline technical parameters, measuring, and calibration regimes can be found elsewhere [31, 32]. When analyzing NEXAFS spectra of organic compounds and polymers, it is necessary to take into account their possible radiation-induced degradation [33]. To exclude this effect, the C K edge and N K edge spectra were repeatedly measured from the same surface area of the samples. The as-measured spectra were virtually unchanged, indicating the radiation stability of the synthesized polyaniline films.
In addition to the NEXAFS spectra, the C1s, N1s, and S2p XPS spectra were acquired. All the XPS spectra were measured with a SPECS PHOIBOS 150 hemispherical energy analyzer. The exciting photon energy was 385.0, 500.0, and 260.0 eV for the C1s, N1s, and S2p spectra, respectively. The energy scale of the spectra was calibrated to take into account the charging effect by referencing the C1s peak of phenyl groups to 284.7 eV [34].
Electrochemical characteristics of the films and composites were measured in 1 M sulfuric acid solution, in which the samples were held for 15 min. After that, the samples were cycled in the range 0–700 mV at different scan rates: 10 cycles at 10 mV s−1, 20 cycles at 20 mV s−1, 50 cycles at 50 mV s−1, 100 cycles at 100 mV s−1, and 1000 cycles at 1000 mV s−1. After the above cycling, the polarization curves were taken again at scan rate 10 mV s−1. A comparison of these curves with those measured at the beginning of the experiment enabled us to evaluate the redox stability of the synthesized films and composites. After thorough mechanical separation from the substrate with a scalpel, the coatings were weighed to evaluate the specific capacitance values. The specific capacitance (c, F g−1) was calculated from the formula:
where ΔQ is voltammetric charge (C) which integrated from cyclic voltammogramm, ΔV is the potential range (V), and m is the mass of film (g) [35].
Results and discussion
Electrodeposition of polyaniline in the presence of sodium dodecyl sulfate and suspended carbon particles and nanotubes
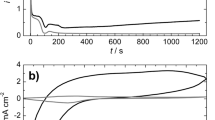
Cyclic voltammograms obtained during polyaniline film formation on graphite are shown in Fig. 1a and b for oxalic acid solutions without and with SDS additives. The voltammograms for the 50th polarization cycle are also shown in Fig. 1c for the films deposited from suspensions of different carbon particles or CNTs. Figure 1d shows the dependence of the polymerization charge density upon cycling for different samples.

Cyclic voltammograms obtained during polyaniline film formation from stirred solution of aniline (0.1 M) and oxalic acid (0.3 M) without (a) and with sodium dodecyl sulfate (0.059 M) additive (b); voltammograms for the 50th cycle under similar conditions with addition of sodium dodecylsulfate and suspension of carbon particles/nanotubes (c). Scan rate 25 mV s−1. Dependence of the anodic charge density on the cycle number for the polyaniline film formation obtained without and with an addition of sodium dodecyl sulfate and carbon particles/nanotubes (d)
The increase of the anodic current observed at the first cycle of polarization starts at the potential above 800 mV, which is associated with the formation of cation radicals of aniline [29, 36, 37]. The coupling of cation radicals results in the formation of oligomers which are then oxidized at lower potentials as compared with the monomer. Anodic oxidation of oligomers leads to the formation of quinonediimine fragments with pronounced oxidizing properties in their protonated form, which anchor aniline molecules (reductant) by predominantly “head-tail” mechanism. This process is then repeated again and facilitates the growth of chains [1, 3, 36, 37]. As compared with sulfuric acid solution [29], the voltammograms of aniline oxidation in oxalic acid solution should be noted to exhibit higher main peak potential and noticeably lower current. The causes are less protonation and, as a result, lower electrical conductivity and electrochemical activity of polyaniline deposited from oxalic acid solution [29].
For the solutions without SDS additive, the current reduces with growing cycle number. Such a behavior is typical for the formation of polyaniline from stirred solutions, including strong acids, owing to relatively little nucleation, and slow growth under these conditions [27]. So, stirring the solution inhibits the aniline oxidation, since catalyzing oligomeric products are taken away from the near-electrode layer. The SDS addition leads to significant growing of the current from cycle to cycle. As the polymer accumulates on the electrode surface, the contribution from pseudocapacitive charging of the already deposited film gradually increases, which also leads to an increase in the anodic current and charge. In addition to these processes, the oxidation of oxalate ions may also give some contribution to the observed anodic currents and charges [29]. By the 50th cycle, the anodic current and charge are approximately an order of magnitude higher for the Pani/SDS as compared with the Pani sample. Similar relationships were obtained for the films grown in the SDS-stabilized suspensions of carbon particles or CNTs.
The effect of SDS on the electrodeposition of polyaniline has been previously studied for quiescent sulfuric acid solution. The accelerating effect of SDS was explained by the accumulation of whether aniline-dodecyl sulfate ion pairs [17] or droplets of SDS-stabilized aniline emulsion [18] at the electrode-solution interface. In the case of oxalic acid solutions, these changes may similarly promote the formation of polyaniline. In addition, it is necessary to take into account a significant increase in the degree of protonation of polymer chains under deposition in the presence of SDS (see below).
For the polyaniline films synthesized from solutions of strong acids, the SDS doping has a relatively weak effect on their protonation and electrical conductivity, and this effect being not always positive [8, 9, 15]. As a result, the role of this factor in the accelerating effect of SDS on the electrodeposition of polyaniline was not previously taken into consideration [17, 18]. The protonation is an extremely important factor due its catalyzing effect on the growth of chains [3, 36] and electrochemical activity of the polymer [28]. In the solutions of oxalic acid being a moderate acid, the difference between the degree of protonation of the chains formed with and without SDS may play a key role in the kinetics and acceleration of the polyaniline electrodeposition. The increase of the current for the films prepared in the presence of suspended carbon particles may be assigned to the increase in the specific surface due to particles capture, as well as faradic and non-faradic processes on their surface.
The SEM micrographs of the films are shown in Fig. 2. The SEM image of the Pani sample taken at high magnification shows that the polymer precipitates in the form of particles of about 300 nm in diameter, which consist, in turn, of smaller polymeric globules. As observed in the image taken at lower magnification, the polyaniline particles are then arranged as larger agglomerates with an extended surface. SDS addition facilitates the formation of polyaniline of a plate-like, not a globular, structure. These plates are organized as large agglomerates of several tens of microns with an extended surface. Similar structures were earlier observed under electrochemical preparation of polyaniline from sulfuric acid solution with a high concentration of SDS [18]. The dimension of such structures was reported [18] to match the characteristic size of droplets of the SDS-stabilized aniline microemulsion.

SEM micrographs of the electrodeposited films surface
The SEM micrographs of the films prepared with suspended carbon particles or CNTs indicate their intensive capture by the growing film. The Pani/SDS_CNT sample is characterized by an extended morphology of the surface with a large number of “tangled” CNTs. The surface is not homogeneous, with particular areas being enriched with the polymer. The Pani/SDS_C A * sample apparently comprises agglomerates of activated graphite particles with thin polymer plates similar to those observed in the Pani/SDS sample.
Chemical structure of electrodeposited PANI films
Figure 3 shows the C1s, N1s, and S2p spectra for Pani and Pani/SDS samples. The main spectral feature in the C1s spectrum at 284.7 eV is due to carbon of benzene and quinone rings, while a shoulder at 286.0 eV arises from the C–N bond [34, 38,39,40]. A shoulder at the binding energy above 287.0 eV may be attributed to various oxygen-containing groups [34], including intercalated oxalate and alkyl sulfate (C–O–S bond) anions, as well as phenolic and quinone derivatives formed by an adventitious oxidation of rings or hydrolysis of imine groups [3]. N1s spectra show the following main features at 398.2 eV from an imine group, 399.3 eV from an amine group, 400.8 eV and 402.3 eV from the protonated imine and amine groups, respectively [40, 41]. An analysis of the N1s spectra shows that the degree of protonation is significantly higher for the Pani/SDS sample than for the Pani sample. For the Pani/SDS sample, not only imine groups but also most of amine groups of the polymer are protonated. A high intensity of the S2p spectrum indicates significant doping of the Pani/SDS sample with dodecyl sulfate anions.

The C1s, N1s, and S2p XPS spectra of the Pani and Pani/SDS films
Figure 4 shows the NEXAFS spectra measured at C K, N K, and S L2,3 edges for the electrodeposited samples. Three characteristic π* resonances are observed in the C K edge spectra. The first resonance of a relatively low intensity located at 284.2 eV is attributed to the transition to the π* orbital of quinone rings. The second one of the highest intensity at 285.3 eV relates to the transition to the π* orbital of benzene rings. An appearance of the third π* resonance at 286.7 eV is caused by the chemical shift for the atoms bound directly with the nitrogen atoms [42]. The peaks in the range of 288.0–289.0 eV are assigned to the σ*(C–H) and 2π* resonances [41,42,43,44,45]. A weak feature at 290.0 eV is due to the C1s→2π*(C–N) transition. Two broad features at 294.0 and 302.0 eV are associated with the σ*(C–C) and σ*(C=C) resonances [42, 43]. The resonances at 397.4 and 402.3 eV in the N K edge spectra are related to the π* resonance of the imine and amine groups, respectively [41,42,43]. The protonated imine group is characterized by the π* resonance at 398.8 eV, while a broad feature at 410.0 eV is associated with the σ* resonances [41,42,43].

C K, N K, and S L2,3 edge NEXAFS spectra measured from the Pani and Pani/SDS films
The C K NEXAFS spectrum from the Pani/SDS sample shows a higher intensity of σ*(C–H) and σ*(C–C) resonances, which are characteristic of alkyl chains [46, 47]. Besides, the S L2,3 NEXAFS spectrum reveals the presence of sulfate groups in the film [48], which confirms the intensive doping of the film with dodecyl sulfate ions. In the N K spectrum measured from the Pani/SDS, there is no observed peak from non-protonated imine groups, indicating almost complete protonation of the film.
The obtained XPS and NEXAFS data allow one to study oxidation state of the deposited polyaniline films. Polyaniline is known to exist in three well distinguishable oxidation states: leucoemeraldine, emeraldine, and pernigraniline. Leucoemeraldine and pernigraniline are the fully reduced (all the nitrogen atoms are of amine) and the fully oxidized (all the nitrogen atoms are of imine) forms, respectively. In emeraldine, the ratio of imine and amine groups is 1:1 [1]. The films were prepared at elevated anodic potentials up to 1200 mV. It should be noted that the formation of pernigraniline is possible even at the potentials exceeding 800 mV [25, 26]. However, the main contributions into the nitrogen XPS and NEXAFS spectra are from the amine groups not containing in the pernigraniline structure, which may indicate its relatively low concentration in the grown films. This can be explained by the effect of stirring which provides the surface with a continuous delivery of aniline as a reducing agent for the pernigraniline fragments formed in the film during its electrochemical oxidation [27, 37].
The formation of a significant amount of leucoemeraldine is also of low probability, since the films were not polarized at the potentials as low as 200 mV, at which emeraldine reduces into leucoemeraldine [25, 26, 37]. The NEXAFS spectra successfully confirm this. The peaks of imine groups (397.4 and 398.8 eV) are observed in the N K edge spectra along with the contributions from the quinone rings (294.2 eV) in the C K edge spectra, with both being not characteristic of leucoemeraldine. Thus, the grown films are of predominantly emeraldine structure. In the case of the Pani sample, the film is rather in some intermediate state between the base and the salt of the emeraldine with relatively low degree of protonation, whereas in the case of the Pani/SDS sample, it is an emeraldine salt intercalated with dodecyl sulfate anions.
The increase in the protonation and electrochemical activity of the films grown with SDS additive may be due to strong interaction of this anion-active surfactant with the cation-active groups of the polymer. Chemical synthesis of polyaniline is often performed with alkyl sulfate additives which significantly increases its protonation and electrical conductivity [10, 42, 49]. Under cyclic polarization, differences in the chemical structure of the polymer can also arise from different mobility of anions in the film, which affects ion exchange needed to compensate the charge and keep electroneutrality [17, 24]. For example, it has been shown for the polyaniline-SDS film electrodeposited in hydrochloric acid solution by quartz crystal microbalance that strong interaction immobilizes the surfactant in the polymer and increases the participation of the solvent molecules during the redox process [17]. Under anodic oxidation at potentials above 200 mV, the amount of ejected protons is lower for the film doped with SDS as compared with the undoped one [17].
The Raman and FTIR spectra of the samples are shown in Figs. 5 and 6. Table 1 presents the assignments of the main bands of the vibrational spectra based on the published data [50,51,52,53,54,55,56,57,58,59,60,61,62]. The Raman spectra measured from the Pani and Pani/SDS samples differ significantly. The strongest band at 1470 cm−1 for the Pani sample is assigned to the stretching vibrations of the C=N bonds [51,52,53,54], which indicates a relatively low degree of protonation of the polymer in this case. For the Pani/SDS film, a strong band at 1340 cm−1 is attributed to the stretching vibrations of the cation radicals. The bands at 745 and 780 cm−1 from the quinone ring deformations [50,51,52] are not observed, indicating a high degree of protonation of the polymer grown in the presence of SDS.

Raman spectra of the Pani and Pani/SDS films, Pani/SDS_CNT, and Pani/SDS_C A * composite films measured from different sites of the surface (#1–#3). The spectra measured from the carbon nanotubes and particles of activated carbon used for synthesizing the films as well as the spectrum of initial non-activated graphite are also shown for comparison. The marked are the main bands (D, G, and D′) from carbon materials and strong δ (C–H) band from polyaniline

FTIR spectra of electrodeposited films all measured in transmission mode, except for the Pani film measured in the reflection mode. Electrodeposition of the Pani film was carried out under the same conditions but onto a polished iron plate
A similar conclusion can be drawn when analyzing the FTIR spectra (Fig. 6). The Pani/SDS spectrum demonstrates stronger absorption at 1140 and 1230 cm−1, characteristic of protonated imine groups [51, 57], as compared with the Pani sample. The Pani/SDS shows several bands from SDS. The quantity of dodecyl sulfate anions can be reliably estimated from the intensity of individual bands of stretching vibrations for alkyl groups at 2850–2960 cm−1.
Figure 5 shows the Raman spectra measured from CNTs and mechanically activated graphite particles used for the film deposition, as well as the spectrum of pristine graphite. The main features in the Raman spectra of carbon particles and CNTs are so-called G and D peaks at around 1580 and 1335 cm−1, respectively. The G peak is due to bonding of all pairs of sp 2 atoms in both rings and chains. The D peak is due to the breathing modes of sp 2 atoms in the rings [63,64,65]. The degree of disorder of the carbon material can be estimated from the ratio between G and D peaks, since it depends on the nanocrystalline domain size: L a=C λ (I D/I G)−1, where L a is the in-plane correlation length and C λ is the coefficient depending on the excitation radiation wavelength (83 Å for λ exc = 632.8 nm) [63,64,65]. Disordering leads to increasing intensity of the D band and broadening the G and D peaks. Also, an additional D′ peak appears at 1615 cm−1. Similar to D band, D′ is a double-resonance Raman feature induced by disordering, defects, or ion intercalation between the graphitic walls [64, 65].
The measured spectra give evidence to some disordering of coarse-grained graphite under ball milling. The most disordered carbon is formed in the ammonium persulfate aqueous solution, which is due to the stronger oxidation of balls and vials accompanied by the apparent intercalation of iron ions and nanosized iron oxides into carbon particles. According to the XPS data, the percentage of iron, mainly in the form of Fe (III) derivatives, is about 1 at.% in the particles. Also, XPS analysis of the particles showed almost complete etching of iron compounds after washing the particles with acid. After acid treatment, the Raman spectra show narrowing the peaks and decreasing the intensity of the D and D′ bands, which indicates some change in the carbon material structure. Based on the Raman spectra, the degree of disorder of carbon materials increase as follows: initial large-crystalline graphite, carbon nanotubes, particles activated in ammonium persulfate aqueous medium after acid treatment, as-synthesized particles activated in heptane, as-synthesized particles activated in ammonium persulfate aqueous medium.
The Raman spectra of the films (Fig. 5) electrodeposited with suspended carbon particles or CNTs were measured from different surface sites. The main bands are the same for carbon materials and polyaniline. The G band matches with the stretch vibrations of the bonds in benzene and quinone rings, while the D band agrees well with the stretch vibrations of cation radicals. Therefore, the percentage of polymer can be easily evaluated based on the intensity of the specific vibration band δ (C–H) at 1165 cm−1. Polyaniline is observed overall the surface, but in different quantities, which indicates the heterogeneity of the chemical composition of the composite films. For all the measured sites of the surface, the spectra are characterized by a relatively low intensity of the band from the stretch vibrations of the C=N bonds at 1470 cm−1, indicating a high degree of protonation of polyaniline in the composite films grown.
The FTIR spectra of the composite films are shown in Fig. 6. The spectra are typical of the polymer, since the main vibrations of graphitic materials are not infrared active [56]. The spectra of the composite films are similar to that of the Pani/SDS sample. Differences are mainly evidenced as different intensities of the bands from dodecyl sulfate anions intercalated into the film. The lowest SDS doping is observed for the film prepared with CNTs, which is evidenced by a very low intensity of the bands of stretching vibrations for alkyl groups (2850–2960 cm−1) in the FTIR spectrum measured from the sample Pani/SDS_CNT. Differences in the SDS doping depended on the type of the captured particles are not yet well understood.
Electrochemical properties of the films
In Fig. 7, voltammograms are shown for the samples measured in 1 M sulfuric acid at the scan rates of 10, 50, and 1000 mV s−1. The potential range of 0–700 mV was chosen because of a relatively high redox stability of polyaniline at these potentials [66]. At higher anodic potentials, polyaniline is prone to overoxidation and hydrolysis, which leads to fast destruction of the polymer and then deteriorating its electrochemical characteristics [25]. Dependences of the charge density on the cycle number and scan rate are shown in Fig. 7d. The presented curves make it also possible to compare the charge for the early stages and after long-term cycling at the same scan rate (10 mV s−1) and then to compare the redox stability of the electrodeposited films.

Stabilized cyclic voltammograms of the films in quiescent solution of sulfuric acid (1 М) measured at scan rates of 10 mV s−1 (a), 50 mV s−1 (b), and 1000 mV s−1 (c). Dependence of the charge density on the cycle number for the films in the 1 M sulfuric acid solution (d). Scan rates are shown in the Figure. The large labels correspond to the charge density at the scan rate of 10 mV s−1 after long-term cycling
The electric current and charge density measured for the Pani/SDS sample are more than an order of magnitude higher than for the Pani sample for all scan rates. This observation is primarily attributed to more intensive deposition of the polymer in the SDS presence and higher protonation of the polymer, which causes its high electrochemical activity. The Pani sample reveals 15% decrease in the charge after its polarization based on the chosen scheme, while the Pani/SDS sample, on the contrary, is characterized by a slight increase in the charge. This result indicates a high redox stability of the dodecyl sulfate-doped polyaniline.
The films grown with SDS additives were weighed to calculate the specific capacitance values. The Pani film was too thin to measure its weight. The calculated values of the specific capacitance of the films are shown in Table 2. The G. Ćirić-Marjanović review includes data on specific capacitance for various composites of polyaniline with carbon particles and nanotubes [4]. Specific capacitance varies over a wide range from 200 to 1500 F g−1 for such composites, depending strongly on the way of their synthesis as well as the measurement conditions. In most cases, the specific capacitance is in the range of 400–600 F g−1, which is close to the values observed in our case. It should be noted that chemical or electrochemical polymerization of aniline is commonly performed in strong acid solutions. In our case, an addition of SDS and carbon materials enabled us to achieve comparable values of specific capacitance for the polymerization of aniline in even oxalic acid solution. Therefore, SDS may be successfully used to deposit polyaniline of high electrochemical activity even from moderate acid solutions. This feature can be used, for example, in the preparation of polyaniline-based composites comprising the particles unstable in strong acidic media, e.g., oxides/oxyhydroxides of some metals.
For the Pani/SDS_CNT sample, the current and charge density are in a good agreement with those for the Pani/SDS. The specific capacitance is 1.5–2 times higher for the Pani/SDS_CNT than for the Pani/SDS (Table 2), which is attributed to relatively low weight of the coating with CNTs which give a great contribution to its extended surface and high electrochemical activity. Another reason for the high electrochemical activity of this composite may also be associated with strong interaction realized often between polyaniline chains and CNTs. Strong π-stacking and electrostatic interaction facilitate the formation of polyaniline with a more planar structure of the chains arranged along the nanotubes, with the increased conductivity and electrochemical activity [23, 59, 67]. Another feature of the Pani/SDS_CNT sample is the increased specific capacitance at high scan rates (Table 2) compared with all other prepared films.
Among the coatings comprising activated graphite particles, the highest current and charge density are observed for the Pani/SDS_C A . This film was characterized by the highest anodic charge under deposition (Fig. 1) and largest weight among all the measured films. Therefore, in the presence of graphite particles not washed in acid and then containing iron compound impurities, the film is grown much faster. The cause of this behavior is not yet clear. Redox reactions of iron compounds accumulated in graphite particles during ball milling in an ammonium persulfate aqueous solution may also make some contribution into increasing the current. It has been previously shown that iron oxide particles play an important role in the electrochemical behavior of graphite particles prepared by ball milling in water [68].
Based on the specific capacitance of the composites, the samples can be arranged in a row: Pani/SDS_C A *>Pani/SDS_C A >Pani/SDS_C H . The differences in the electrochemical activity of the samples may arise from different chemical and/or structural state of graphite particles. The XPS analysis of the particles showed the same quantity of oxidized carbon in all cases (not higher than 5 at.%) and relatively low percentage of iron impurities (about 1 at.%) for the Pani/SDS_C A sample. This makes it possible to exclude significant effect of the chemical composition of the particles on the electrochemical activity of the composite films.
The structural state of the particles differs more significantly, as shown by the Raman spectra (Fig. 5). However, there is no straightforward correlation between the degree of disorder in the particles and the electrochemical activity of the composites based on these particles. Maximum activity is observed for the Pani/SDS_C A * sample containing the particles with the most ordered structure. Apparently, similar to the case of CNTs, some interaction (π-stacking and/or electrostatic) of polyaniline with the surface of graphite particles can be assumed, which contributes to the formation of more planar structure of the polymer and then its higher conductivity and electrochemical activity. With an accumulation of great quantity of defects in the carbon material, such interaction is likely weakened and the electrochemical activity of the polymer decreases. This effect is possibly observed in the case of Pani/SDS_C A and Pani/SDS_C H composites comprising carbon particles of the most disordered structure. Unlike the Pani/SDS sample, all the electrodeposited composite coatings demonstrate a slight degradation under long-term cycling, resulting in 7–10% reduction of the charge density (Fig. 7).
Conclusion
It has been shown that SDS additives significantly accelerate the PANI electrodeposition in oxalic acid solution. The as-formed PANI is doped with dodecyl sulfate ions and characterized by protonation of all imine groups and large amount of amine groups. SDS additives facilitate the formation of PANI of plate-like structure with an extended surface. The changes in the chemical structure and morphology provide a significant increase in the electrochemical activity of PANI at high redox stability. An introduction of carbon particles or CNTs into the electrolyte in addition to SDS leads to intensive capture of suspended particles by the polymer under its growth and then the formation of composite coatings. The electrochemically deposited composite coatings are characterized by an extended surface and high specific capacitance values. Owing to the revealed features of the structure and properties, the as-synthesized coatings are considered as promising material for their use in various electrochemical devices (capacitors, sensors, etc.).
References
Gospodinova N, Terlemezyan L (1998) Conducting polymers prepared by oxidative polymerization: polyaniline. Prog Polym Sci 23:1443–1484
Bhadra S, Khastgir D, Singha NK, Lee JH (2009) Progress in preparation, processing and applications of polyaniline. Prog Polym Sci 34:783–810
Ćirić-Marjanović G (2013) Recent advances in polyaniline research: polymerization mechanisms structural aspects, properties and applications. Synthetic Met 177:1–47
Ćirić-Marjanović G (2013) Recent advances in polyaniline composites with metals, metalloids and nonmetals. Synthetic Met 170:31–56
Tallman DE, Spinks G, Dominis A, Wallace GG (2002) Electroactive conducting polymers for corrosion control. Part 1. General introduction and a review of non-ferrous metals. J Solid State Electrochem 6:73–84
Spinks G, Dominis A, Wallace GG, Tallman DE (2002) Electroactive conducting polymers for corrosion control. Part 2. Ferrous metals. J Solid State Electrochem 6:85–100
Ates M, Sezai Sarac A (2009) Conducting polymer coated carbon surfaces and biosensor applications. Prog Org Coat 66:337–358
Hino T, Namiki T, Kuramoto N (2006) Synthesis and characterization of novel conducting composites of polyaniline prepared in the presence of sodium dodecylsulfonate and several water soluble polymers. Synthetic Met 156:1327–1332
Stejskal J, Omastová M, Fedorova S, Prokeš J, Trchová M (2003) Polyaniline and polypyrrole prepared in the presence of surfactants: a comparative conductivity study. Polymer 44:1353–1358
Kuramoto N, Geniès EM (1995) Micellar chemical polymerization of aniline. Synthetic Met 68:191–194
Kim B-J, Oh S-G, Han M-G, Im S-S (2000) Preparation of polyaniline nanoparticles in micellar solutions as polymerization medium. Langmuir 16:5841–5845
Leng W, Zhou S, Gu G, Wu L (2012) Wettability switching of SDS-doped polyaniline from hydrophobic to hydrophilic induced by alkaline/reduction reactions. J Colloid Interf Sci 369:411–418
Leng W, Zhou S, You B, Wu L (2012) Controllable synthesis of aniline oligomers into uniform, dispersed cross and needle morphologies. J Colloid Interf Sci 374:331–338
Rahmanifar MS, Mousavi MF, Shamsipur M, Riahi S (2006) A study on the influence of anionic surfactants on electrochemical degradation of polyaniline. Polym Degrad Stabil 91:3463–3468
Cai L-T, Yao S-B, Zhou S-M (1997) Surfactant effects on the polyaniline film. Synthetic Met 88:209–212
Michaelson JC, McEvoy AJ, Kuramoto N (1992) Morphology and growth rate of polyaniline films modified by surfactants and polyelectrolytes. React Polym 17:197–206
De Albuquerque Maranhão SL, Torresi RM (1999) Quartz crystal microbalance study of charge compensation process in polyaniline films doped with surfactant anions. Electrochim Acta 44:1879–1885
Kanungo M, Kumar A, Contractor AQ (2002) Studies on electropolymerization of aniline in the presence of sodium dodecylsulfate and its application in sensing urea. J Electroanal Chem 528:46–56
Prathap MUA, Thakur B, Sawant SN, Srivastava R (2012) Synthesis of mesostructured polyaniline using mixed surfactants, anionic sodium dodecylsulfate and non-ionic polymers and their applications in H2O2 and glucose sensing. Colloid Surface B 89:108–116
Herrasti P, Recio FJ, Ocón P, Fatás E (2005) Effect of the polymer layers and bilayers on the corrosion behaviour of mild steel: comparison with polymers containing Zn microparticles. Prog Org Coat 54:285–291
Chang G, Luo Y, Liao F, Lu W, Sun X (2011) Polyacetylene nanoparticles-based preparation of polyaniline nanofibers. J Nanopart Res 13:471–477
Vaidya SG, Rastogi S, Aguirre A (2010) Surfactant assisted processable organic nanocomposite dispersions of polyaniline–single wall carbon nanotubes. Synthetic Met 160:134–138
Oueiny C, Berlioz S, Perrin F-X (2014) Carbon nanotube–polyaniline composites. Prog Polym Sci 39:707–748
De Albuquerque Maranhão SL, Torresi RM (1999) Anion and solvent exchange as a function of the redox states in polyaniline films. J Electrocem Soc 146:4179–4182
Orata D, Buttry DA (1987) Determination of ion population and solvent content as function of redox state and pH in polyaniline. J Am Chem Soc 109:3574–3581
Horanyi G, Inzelt G (1988) Anion-involvement in electrochemical transformation of polyaniline. A radiotracer study. Electrochim Acta 33:947–952
Сórdova R, Del Valle MA, Arratia A, Gómez H, Schrebler R (1994) Effect of anions on the nucleation and growth mechanism of polyaniline. J Electroanal Chem 377:75–83
Duić L, Mandić Z (1992) Counter-ion and pH effect on the electrochemical synthesis of polyaniline. J Electroanal Chem 335:207–221
Erdem E, Saçak M, Karakişla M (1996) Synthesis and properties of oxalic acid-doped polyaniline. Polym Int 39:153–159
Makarova TL, Zakharchuk I, Geydt P, Lahderanta E, Komlev AA, Zyrianova AA, Lyubchyk A, Kanygin MA, Sedelnikova OV, Kurenya AG, Bulusheva LG, Okotrub AV (2016) Assessing carbon nanotube arrangement in polystyrene matrix by magnetic susceptibility measurements. Carbon 96:1077–1083
Gorovikov SA, Follath R, Molodtsov SL, Kaindl G (2001) Optimization of the optical design of the Russian–German soft-X-ray beamline at BESSY II. Nucl Instrum Meth A 467-468:565–568
Watts B, Thomsen L, Dastoor PC (2006) Methods in carbon K-edge NEXAFS: experiment and analysis. J Electron Spectrosc Relat Phenom 151:105–120
Coffey T, Urquhart SG, Ade H (2002) Characterization of the effects of soft X-ray irradiation on polymers. J Electron Spectrosc Relat Phenom 122:65–78
Beamson G, Briggs D (1992) High resolution XPS of organic polymers. The Scienta ESCA300 database. John Wiley & Sons, New York
Ates M, Akif Serin M, Ekmen I, Ertas YN (2015) Supercapacitor behaviors of polyaniline/CuO, polypyrrole/CuO and PEDOT/CuO nanocomposites. Polym Bull 72:2573–2589
Yang H, Bard AJ (1992) The application of fast scan cyclic voltammetry. Mechanistic study of the initial stage of electropolymerization of aniline in aqueous solutions. J Electroanal Chem 339:423–449
Duić L, Mandić Z, Kovać S (1995) Polymer-dimer distribution in the electrochemical synthesis of polyaniline. Electrochim Acta 40:1681–1688
Wei X-L, Fahlman M, Epstein AJ (1999) XPS study of highly sulfonated polyaniline. Macromolecules 32:3114–3117
Zeng X-R, Ko T-M (1998) Structures and properties of chemically reduced polyanilines. Polymer 39:1187–1195
Neoh KG, Kang ET, Tan KL (1991) Structural study of polyaniline films in reprotonation/deprotonation cycles. J Phys Chem 95:10151–10156
Hennig C, Hallmeier KH, Szargan R (1998) XANES investigation of chemical states of nitrogen in polyaniline. Synthetic Met 92:161–166
Magnuson M, Guo J-H, Butorin SM, Agui A, Såthe C, Nordgren J, Monkman AP (1999) The electronic structure of polyaniline and doped phases studied by soft X-ray absorption and emission spectroscopies. J Chem Phys 111:4756–4761
Guay D, Stewart-Ornstein J, Zhang X, Hitchcock AP (2005) In situ spatial and time-resolved studies of electrochemical reactions by scanning transmission X-ray microscopy. Anal Chem 77:3479–3487
Yau S, Lee YH, Chang CZ, Fan LJ, Yang YW, Dow WP (2009) Structures of aniline and polyaniline molecules adsorbed on Au (111) electrode: as probed by in situ STM, ex situ XPS, and NEXAFS. J Phys Chem C 113:13758–13764
Lee YH, Chang CZ, Yau SL, Fan LJ, Yang YW, Ou Yang LY, Itaya K (2009) Conformations of polyaniline molecules adsorbed on Au(111) probed by in situ STM and ex situ XPS and NEXAFS. J Am Chem Soc 131:6468–6474
Fu J, Urquhart SG (2005) Linear dichroism in the X-ray absorption spectra of linear n-alkanes. J Phys Chem A 109:11724–11732
Hähner G, Zwahlen M, Caseri W (2005) Solvent dependence of the molecular order in ion-exchanged self-assembled dialkylammonium monolayers on mica studied with soft X-ray absorption. J Colloid Interf Sci 291:45–52
Jalilehvand F (2006) Sulfur: not a “silent” element any more. Chem Soc Rev 35:1256–1268
Adams PN, Laughlin PJ, Monkman AP, Kenwright AM (1996) Low temperature synthesis of high molecular weight polyaniline. Polymer 37:3411–3417
Colomban P, Folch S, Gruger A (1999) Vibrational study of short-range order and structure of polyaniline bases and salts. Macromolecules 32:3080–3092
Trchová M, Morávková Z, Bláha M, Stejskal J (2014) Raman spectroscopy of polyaniline and oligoaniline thin flms. Electrochim Acta 122:28–38
Ćirić-Marjanović G, Trchová M, Stejskal J (2008) The chemical oxidative polymerization of aniline in water: Raman spectroscopy. J Raman Spectrosc 39:1375–1387
Louran G, Lapkowski M, Quillard S, Pron A, Buisson JP, Lefrant S (1996) Vibrational properties of polyaniline—isotope effect. J Phys Chem 100:6998–7006
Quillard S, Louarn G, Lefrant S, Macdiarmid AG (1994) Vibrational analysis of polyaniline: a comparative study of leucoemeraldine, emeraldine, and pernigraniline bases. Phys Rev B 50:12496–12508
D’Antonio MC, Wladimirsky A, Palacios D, Coggiola L, González-Baró AC, Baran EJ, Mercader RC (2009) Spectroscopic investigations of iron(II) and iron(III) oxalates. J Braz Chem Soc 20:445–450
Mayo DW, Miller FA, Hannah RW (2003) Course notes on the interpretation of infrared and Raman spectra. John Wiley & Sons Inc, Hoboken
Kellenberger A, Dmitrieva E, Dunsch L (2011) The stabilization of charged states at phenazine-like units in polyaniline under p-doping: an in situ ATR-FTIR spectroelectrochemical study. Phys Chem Chem Phys 13:3411–3420
Chatterjee S, Salaün F, Campagne C (2014) The influence of 1-butanol and trisodium citrate ion on morphology and chemical properties of chitosan-based microcapsules during rigidification by alkali treatment. Mar Drugs 12:5801–5816
Salvatierra RV, Oliveira MM, Zarbin AJG (2010) One-pot synthesis and processing of transparent, conducting, and free standing carbon nanotubes/polyaniline composite films. Chem Mater 22:5222–5234
Šeděnková I, Trchová M, Stejskal J (2008) Thermal degradation of polyaniline films prepared in solutions of strong and weak acids and in water—FTIR and Raman spectroscopic studies. Polym Degrad Stabil 93:2147–2157
Nakanishi K (1962) Infrared absorption spectroscopy. Holden-Day Inc. San Francisco & Nankodo Company Limited, Tokyo
NIST Chemistry WebBook (2011) National Institute of Standards and Technology, Gaithersburg. http:webbook.nist.gov/chemistry. Accessed 1 June 2017
Ferrari AC (2007) Raman spectroscopy of graphene and graphite: disorder, electron–phonon coupling, doping and nonadiabatic effects. Solid State Commun 143:47–57
Osswald S, Havel M, Gogotsi Y (2007) Monitoring oxidation of multiwalled carbon nanotubes by Raman spectroscopy. J Raman Spectrosc 38:728–736
Bokobza L, Bruneel J-L, Couzi M (2015) Raman spectra of carbon-based materials (from graphite to carbon black) and of some silicone composites. C 1:77–94
Wu M, Snook GA, Gupta V, Shaffer M, Fray DJ, Chen GZ (2005) Electrochemical fabrication and capacitance of composite films of carbon nanotubes and polyaniline. J Mater Chem 15:2297–2303
Huang J-E, Li X-H, Xu J-C, Li H-L (2003) Well-dispersed single-walled carbon nanotube/polyaniline composite films. Carbon 41:2731–2736
Janot R, Guérard D (2002) Ball-milling: the behavior of graphite as a function of dispersal media. Carbon 40:2887–2896
Acknowledgements
We are thankful to Dr. S.F. Lomayeva (Physical-Technical Institute, UB RAS, Izhevsk, Russia) for ball milling of graphite, Dr. K.G Mikheev (Institute of Mechanics, UB RAS, Izhevsk, Russia) for measuring Raman spectra and Dr. D.A. Smirnov (Institut für Festkörperphysik, Technische Universität Dresden) for his technical assistance during our beamtime.
Funding
This work was supported by FASO of Russia within the state assignment No. АААА-А17-117022250038-7, the Russian Foundation for Basic Research (projects No. 16-33-00190 and No. 16-43-180228), and bilateral Program “Russian-German Laboratory at BESSY II”.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Syugaev, A.V., Lyalina, N.V., Maratkanova, A.N. et al. Effect of sodium dodecyl sulfate and carbon particles/nanotubes on electrodeposition of polyaniline from oxalic acid solution. J Solid State Electrochem 22, 931–942 (2018). https://doi.org/10.1007/s10008-017-3822-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-017-3822-x