
Abstract
Since their market introduction in 1991, lithium ion batteries (LIBs) have developed evolutionary in terms of their specific energies (Wh/kg) and energy densities (Wh/L). Currently, they do not only dominate the small format battery market for portable electronic devices, but have also been successfully implemented as the technology of choice for electromobility as well as for stationary energy storage. Besides LIBs, a variety of different technologically promising battery concepts exists that, depending on the respective technology, might also be suitable for various application purposes. These systems of the “next generation,” the so-called post-lithium ion batteries (PLIBs), such as metal/sulfur, metal/air or metal/oxygen, or “post-lithium technologies” (systems without Li), which are based on alternative single (Na+, K+) or multivalent ions (Mg2+, Ca2+), are currently being studied intensively. From today’s point of view, it seems quite clear that there will not only be a single technology for all applications (technology monopoly), but different battery systems, which can be especially suitable or combined for a particular application (technology diversity). In this review, we place the lithium ion technology in a historical context and give insights into the battery technology diversity that evolved during the past decades and which will, in turn, influence future research and development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
One of today’s most challenging issues of mankind is the preservation of a consistent energy supply that is able to meet the world’s increasing energy demands. The development of novel technologies is of utmost importance to ensure sustainable long-term energy generation, conversion and storage. The present “energy economy” is considered to be at serious risk as it is still and to a large extent depending on fossil fuels. This risk concern gives rise to the development of renewable energies such as wind or solar power. This trend is not only due to the increasing shortages of non-renewable (fossil) resources, but also related to the growing concerns about the environmental impact of fossil fuel combustion products including global warming and (air) pollution. Beijing has become famous as just one representative for a vast number of metropolitan cities where the people strongly suffer from the high air pollution by smoke and fog, better known as smog. It has been known for quite some time that air pollutants such as ozone or fine dust particles are harmful to health. According to the most recent estimates of the international energy agency (IEA), more than six million people worldwide die from the consequences of combustion exhaust gases per year [1].
One major strategy to tackle these immense problems lies in the integration of clean and efficient energy storage from renewables into different energy sectors such as transportation and stationary storage. Electrochemical energy storage in the form of rechargeable batteries is the most efficient and feasible solution for various types of storage applications, for small-scale as well as large-scale utilization. The lithium ion technology revolutionized energy storage since its market introduction in 1991 [2], while an evolutionary development with continuously increasing energy contents took place in the recent decades, as reported in various reviews [3,4,5,6,7,8,9,10,11,12,13,14,15,16,17]. Currently, lithium ion batteries (LIBs) do not only dominate the small format battery market for portable electronic devices (laptops, cell phones, etc.), but have also been successfully implemented as the technology of choice for automotives like hybrid (HEV), plug-in (PHEV), or fully electric vehicles (BEV) as well as for stationary energy storage [15, 18, 19].
Besides the state-of-the-art LIBs, a variety of different technologically promising battery chemistry approaches exists depending on the respective storage technology that it might also be suitable for either automotive or stationary application purposes. These systems of the “next generation,” the so-called post-lithium ion batteries (PLIBs), such as metal-sulfur, metal-air or metal-oxygen as well as systems based on alternative single (Na+, K+) or multivalent ions (Mg2+, Ca2+), i.e., post-Li battery systems, are currently intensively studied [20,21,22,23,24,25,26]. The authors of this review will use the terms “next-generation battery”, “beyond-lithium ion battery”, “post-lithium ion battery (PLIB)” as well as “post-lithium battery” in consideration, that the progress of these related alternative battery systems is compared with the already well-performing state-of-the-art LIB technology and can be realized successfully in the future, i.e., after (= post, beyond, or next) the successful realization of the LIB. It is today however, uncertain whether the alternative battery systems will sustainably succeed, thus, will ever (partially) replace the LIB. Any roadmap on battery development should regard this uncertainty, as well as that it is the reality that we have many battery chemistries used in parallel already today and that there is no reason to believe, expect or hope that this will be different in the future.
Another next-generation battery approach is aiming at the so-called “all-solid-state battery” (ASSB), which utilizes a solid electrolyte (SE) and recently raised enormous expectations with regard to operational safety, flexible cell geometry as well as high energy density [27, 28]. In particular, there is a strong academic and industrial interest in an in-depth evaluation and comparison of these different battery systems in terms of theoretical and practical specific energy (Wh kg−1) and energy density (Wh L−1) as well as costs [29,30,31,32,33,34,35]. A major focus lies on the evaluation of the practical specific energy and energy density from the material to the system level of the PLIBs and their potential to reach commercialization in order to replace the state-of-the-art LIBs. Among these various technologies, the sodium ion and lithium/sulfur battery systems are rated to have the highest potential for commercialization in the near future [33].
In this paper, we place the lithium ion technology in a historical context and give insights into the battery technology diversity that evolved during the past decades, as there will be more than just one technology to power all the various applications.
Historic flashback on battery development
From aqueous to non-aqueous battery systems
Figure 1 shows a timeline of selected important discoveries and developments of primary and secondary battery technologies from 1800 until today. The early works of Luigi Galvani (University of Bologna, Italy) and Alessandro Volta (University of Pavia, Italy) at the end of the eighteenth century led to the discovery of the first electrochemical cell. In his experiments, Galvani could observe the muscle twitch of a frog’s leg when touched by a series of two different metallic electrodes. While Galvani believed that animals could generate electricity, Volta recognized that the reason for the muscle contraction was due to the voltage generated by the two different metals. Based on these considerations, Volta developed the first electrochemical cell consisting of zinc and copper plates which were separated by cardboard or leather spacers soaked with an acidic electrolyte, which was later named “Volta pile” (1800). Different electrochemical reactions could take place during the discharge reaction of the Zn/Cu element, depending on whether the system was in contact to atmospheric oxygen. If the system was not closed to the atmosphere, the atmospheric oxygen reacted with copper to form copper(II) oxide (CuO) at the surface of the metal plate. The cell could produce an electric current by oxidation of Zn and reduction of CuO to copper at a cell voltage of 1.1 V (according to Nernst). Accordingly, as oxygen is necessary for the formation of CuO, this first primary battery can also be considered as metal/air system. In contrast, if no oxygen was present, the formation of CuO could not occur, which resulted in a different electrochemical reaction at the cathode, namely the generation of hydrogen gas, at a cell voltage of only 0.76 V (according to Nernst) [36,37,38].

Battery development from 1800 until today showing selected important discoveries of primary and secondary (rechargeable) battery technologies
In 1802, the German physicist Johann Wilhelm Ritter was on the track for a new discovery: He developed a cell that consisted of a glass tube which was filled with a saline solution and closed by corks on both sides, each equipped with gold wires. Ritter used the Volta pile to charge his cell. When a current was applied through the glass tube, he could observe the generation of gas bubbles at both wires, which were later discovered as oxygen on the one side and hydrogen on the other side. By this experiment, Ritter showed the first accumulator principle, a rechargeable battery, in which the electrolysis of water took place during charge and the recombination of oxygen and hydrogen to water occurred during discharge. In addition to the development of the first rechargeable battery, Ritter designed the first galvanic cell, based on the redox reaction of H2 with O2, which can be considered as the forerunner of the fuel cell [38].
These early discoveries were followed by the subsequent development of further primary and secondary battery technologies using aqueous electrolytes. Wilhelm Josef Sinsteden, a German physicist and physician, invented the rechargeable lead-acid battery in 1854. In 1859, Gaston Raimond Planté, a French physicist and paleontologist, developed an improved construction leading to the first commercially viable and today well-known lead-acid battery, which nowadays still dominates the sector of starter and backup batteries, despite its relatively low practical specific energy (20–40 Wh kg−1) [30, 39].
Another primary battery system was invented by the French engineer Georges-Lionel Leclanché in 1866. This battery was based on a zinc rod as negative electrode and a manganese dioxide (MnO2)-carbon mixture as positive electrode using an aqueous electrolyte consisting of ZnCl2 and NH4Cl. Since the 1960s, the original technology was redesigned by changing the acidic electrolyte to a much better conducting alkaline electrolyte of aqueous potassium hydroxide (KOH). Nowadays, these primary batteries, the so-called universal alkaline batteries, are still being used for many small-scale applications [36, 39]. Another primary battery technology, which based on zinc as negative electrode material, was the zinc-mercury oxide (Zn-HgO) battery using an alkaline electrolyte (KOH). This technology was known as Ruben-Mallory (RM) battery, named after the inventor Samuel Ruben and the leading manufacturer P. R. Mallory and Co Inc. (today: Duracell), and was introduced during the 1960s to power the first pacemakers [40]. However, the battery lifetime amounted only for about 2 years, which is too short for pacemaker applications. Today, this type of battery is not being used anymore to avoid the usage of toxic mercury, and, more likely, as it was replaced by primary lithium batteries which showed a remarkably enhanced energy content and lifetime.
Nickel-based systems constitute another important type of battery technology. While the nickel-cadmium battery (Ni-Cd) invention was finally credited to the Swedish engineer Waldemar Jungner in 1899, the nickel-iron (Ni-Fe) battery invention was ascribed by Thomas Edison in 1901. Both systems were the forerunners of the nickel-metal hydride (Ni-MH) battery, which was commercialized in 1989 and showed an improved practical specific energy (70–100 Wh kg−1) compared to the Ni-Cd and Ni-Fe batteries (20–50 Wh kg−1) [39]. Since its market introduction, the Ni-MH battery was largely used to power small-scale consumer electronics and was also applied as the system of choice for many of Toyota’s hybrid electric vehicles such as the Toyota Prius [30]. However, in the recent decades, the nickel-based systems have been more and more replaced by higher energy density batteries, namely LIBs, using—just like the Ni-MH battery—an ion transfer mechanism between the two electrodes.
In 1938, the first rechargeable ion transfer cell consisting of two graphite electrodes and an aqueous electrolyte, namely concentrated sulfuric acid, was published by Rüdorff and Hofmann [41]. In this type of cell, HSO4 − anions are shuttled between the two host electrodes during charge and discharge (Fig. 2).

HSO4 − ion transfer cell composed of two graphite electrodes and concentrated sulfuric acid as electrolyte (one graphite electrode is put in the cell in the intercalated state, thus contains HSO4 - anions, while the other graphite electrode is assembled free of ions). a Charge process: HSO4 − anions are intercalated into the positive graphite electrode, while the negative graphite electrode remains free of ions. b Discharge process: HSO4 − anions within the positive graphite electrode are transferred to and intercalated into the negative graphite electrode
Therefore, this anion ion transfer cell can be considered as the earliest ancestor of the lithium ion transfer system, the so-called lithium “rocking chair” battery, that is based on the same working principle, i.e., the transfer of ions (here: lithium ions) between two insertion electrodes during the charge/discharge process. Furthermore, Rüdorff and Hofmann developed the first rechargeable battery based on graphitic carbon intercalation electrodes; graphite is currently being used as state-of-the-art anode material in LIBs. In 1989, a rechargeable battery with an alternative charge/discharge mechanism, which uses two graphite electrodes in combination with a non-aqueous electrolyte, was introduced by McCullough et al. [42,43,44,45] and further studied by Carlin et al. [46, 47] during the 1990s and by Seel and Dahn in 2000 [48, 49]. This system, the so-called dual-carbon or dual-graphite cell, which enables cell voltages of up to 5 V, is based on the simultaneous intercalation of the electrolyte cations into the graphite anode and of the electrolyte anions into the graphite cathode during charge. Thus, the storage mechanism in this type of cell varies from the ion transfer mechanism, where the electrolyte basically serves only as the ion transfer medium. Here, the electrolyte has to be considered as active material and will have a huge impact on the practical energy density and the cell design, in particular on the electrolyte layer thickness. In order to differentiate this storage mechanism from the classical ion transfer mechanism, Placke et al. introduced the term dual-ion cell in 2012 [50, 51]. Nowadays, dual-ion cells—or their specific example, dual-graphite cells—are seriously considered as an alternative to the lithium ion batteries [52,53,54,55,56,57], even though they cannot compete in terms of specific energy or energy density, but show environmental, safety, and cost benefits and, thus, are an interesting option for stationary energy storage. This technology has not been commercialized yet, even though the start-up company Power Japan Plus announced plans to commercialize this battery system [58], named Ryden battery, in 2014.
From primary lithium metal batteries to secondary lithium ion batteries
In the late 1960s, many technological innovations were made, which resulted in various novel battery technologies. This progress was most likely promoted by the increasing demand for portable energy in many different sectors, particularly including implantable medical devices, military applications, and consumer electronics. Thereby, it was realized that the conventional aqueous battery systems could not cope with the increasing demand of energy, as these batteries were simply too large and too heavy to satisfactorily serve the abovementioned applications [36].
The development of a new concept of batteries using metallic lithium as negative electrode in combination with non-aqueous electrolytes brought the technological breakthrough by delivering remarkably enhanced specific energies and energy densities. The fundamental advantage of using metallic lithium is the low equivalent weight, the correspondingly high specific, and volumetric capacities (3860 mAh g−1, 2047 mAh cm−3) as well as the very low electrode potential of −3.05 V (vs. standard hydrogen electrode), which in turn allows high cell voltages with appropriate cathodes [59]. A further major advantage of lithium metal is its kinetic stability in various non-aqueous (aprotic) electrolytes. Despite the thermodynamic instability of the electrolyte, an electronically insulating passivation layer is formed by initial electrolyte decomposition on its surface. This so-called solid electrolyte interphase (SEI) protects the electrolyte from further degradation, but, in an ideal case, is permeable for the transport of lithium ions. The protective SEI layer, which is also formed on various anode materials for lithium ion batteries that operate at potentials typically below 1 V vs. Li/Li+ (depending on the electrolyte components), critically determines the long-term stability, calendar life, and safety of the battery, and has been intensively studied in the last decades [60,61,62,63,64,65,66,67,68].
A vast number of primary lithium metal-based cell technologies were developed in the 1970s including a broad range of different cathode materials such as iodine (I2), manganese dioxide (MnO2), thionyl chloride (SOCl2), sulfur dioxide (SO2), copper oxide (CuO), carbon monofluoride (CF x ), silver vanadium oxide (SVO; Ag2V4O11), pyrite (FeS2), copper sulfide (CuS), vanadium pentoxide (V2O5), silver chromate (Ag2CrO4), and many more [69,70,71,72]. An overview of the most important primary battery systems, including their practical specific energies and energy densities, is given in Table 1. In 1972, the Japanese company Matsushita Electric Industrial Co. Ltd. developed the Li/CF x battery [73], which was used for aerospace as well as pacemaker applications. Indeed, many primary lithium batteries have been developed for utilization in the medical sector. One of the most famous battery systems is the lithium-iodine cell, which was patented by James R. Moser and Alan A. Schneider in 1972 [74]. The Li/I2 system, which is able to deliver a constant and continuous current in the microampere range, displays a long lifetime of about 10 years and is, thus, nowadays still used as pacemaker battery and for other implantable devices [37]. Another key driving force for the development of primary lithium batteries was the market introduction of a series of consumer electronics in the 1970s, such as electronic watches and cameras. Many of the applications could be satisfied by the Li/MnO2 battery, which was fabricated as a coin-type cell and, therefore, could fulfill the requirements for a small volume size [36].
The success of the primary lithium batteries stimulated an obvious interest to develop secondary, rechargeable battery technologies. In this context, the attention was initially focused on the development of novel cathode materials which were able to take up and release the electrochemically active species inside a host material. In the late 1970s, the breakthrough was achieved by discovery of the so-called “intercalation” or “insertion” materials, such as tantalum disulfide (TaS2) or titanium disulfide (TiS2). TiS2, as the first intercalation cathode, was discovered by Whittingham while he was working at Exxon Enterprises Battery Division, who, in turn, manufactured the first rechargeable lithium battery based on this material in 1976–78 [77,78,79]. This system, using either a metallic lithium or LiAl anode, appeared attractive because of the “high” operating voltage of 2.2 V, which led to a specific energy of ≈40–45 Wh kg−1, as well as the full range of intercalation (0 < x < 1) without a phase transformation for Li x TiS2 [80]. In addition, four further commercial rechargeable lithium metal batteries namely Li/MoS2 (Moli Energy [81, 82]), Li/V2O5 (Toshiba [75]), Li/V3O8 (cell operating at 80–120 °C and based on a polymer electrolyte [30, 83]), and Li/MnO2 (Tadiran [84, 85]) were introduced.
However, due to major safety concerns, the commercial breakthrough of rechargeable batteries based on lithium metal failed and led to their withdrawal from the market [72, 86]. These safety risks were clearly associated with the anode: During the charge process in liquid organic electrolytes, metallic lithium is not deposited homogeneously, but in form of high surface area lithium (HSAL), often referred to as “dendrites,” which can have different structures, such as needle-like (= dendritic), mossy, or granular deposits [87, 88]. This process not only consumes electrolyte by continuous re-formation of the SEI layer in each cycle, but also results in high safety risks (i) because of the high reactivity of HSAL with the electrolyte and (ii) especially when the dendrites grow from the anode towards the cathode and cause a short circuit, which, in turn, can cause fire and even explosion of the cell. In contrast, Li/MnO2 batteries contained an internal safety system, which could maintain a high cycle life of the Li metal anode. Nevertheless, this system was also a commercial failure because cycle life was limited as continuous reaction of the Li metal electrode with the electrolyte led to drying out of the electrolyte and the cell required very long charging times in the range of several hours [11, 84, 85]. Although the use of lithium metal anodes in rechargeable batteries clearly offers advantages in terms of specific energies and energy densities, their broad commercial use in batteries could not be established up to now. Nevertheless, today the rechargeable Li metal anode is even considered as the “holy grail” for lithium battery research and development, which particularly includes the lithium metal-based post-lithium ion technologies (e.g., lithium/air, lithium/sulfur, or ASSBs) [11, 30].
On the route to today’s lithium ion battery, it was clear that the lithium metal anode had to be replaced by a more reliable and safe anode material. Hereby, the new charge/discharge concept considered the combination of two host electrodes which were able to reversibly accept and release lithium ions. In this approach, called first “rocking chair” battery [89, 90], later on lithium ion cell, the negative electrode active material acts the “lithium sink” and the positive electrode active material as the “lithium source” during charge, while the opposite situation is found during discharge of the cell [36, 87]. Therefore, the major difference between lithium metal cells and lithium ion cells was not only the introduction of an anode host material for lithium ions, but also the fact that a cathode material, which had to act as the lithium source during charge, needed to be found. In 1980, Goodenough and collaborators discovered a new class of cathode materials, the layered transition-metal oxides such as LiCoO2 [91], which later on was found to be a breakthrough for the development of today’s LIB. The search for a suitable anode material finally led to the discovery of carbonaceous materials such as graphitic or amorphous carbons, which showed the ability to take up lithium ions by intercalation or insertion [92, 93]. This discovery was based on the early studies on lithium-graphite intercalation compounds (Li x C6) by chemical and electrochemical means [94,95,96,97,98,99,100,101, 105], while the electrochemical intercalation of lithium into graphite was presented by Besenhard in 1976 [101]. The first (intended) use of carbons as anode material in LIBs was reported by the Bell Telephone Laboratories in 1981 [106]. Overall, the carbonaceous materials displayed only a minor volume changes upon lithiation and had a potential close to the one of metallic lithium, which is preferable for achieving a high energy density. In combination with a suitable electrolyte, in most cases carbonate-based electrolytes, the carbons showed a high reversibility for lithium uptake and release. Finally, it should be noted, that a major difference between Li metal batteries and LIBs is that LIBs are assembled in the discharged state. Therefore, they need “formation,” i.e., during the first charge process, the cell is “activated” and an appropriate SEI is formed on the anode, as well as a CEI (cathode electrolyte interphase) [102,103,104, 107] on the cathode. This process is carried out by the cell manufacturer, as it requires significant experience and knowledge to initiate a proper activation, as well as create SEI and CEI with desired properties.
In 1991, the commercial breakthrough of the lithium ion technology was achieved by Sony Corporation’s introduction of a high energy (80 Wh kg−1; 200 Wh L−1) and high voltage (≈3.7 V) cell based on coke as anode material, a non-aqueous electrolyte and LiCoO2 as cathode material [2, 108, 109]. Even though the general concept of two insertion electrodes with largely different redox potentials for Li insertion is still state-of-the-art for today’s LIBs, there has been a continuous development in terms of advanced components considering the active (anode, cathode) and inactive materials (electrolyte, separator, binder, conductive additive, etc.) as well as improvements in manufacturing and engineering, which resulted in a steady, stepwise increase of the lithium ion cell’s specific energies and energy densities in the last decades (Fig. 3). Nevertheless, today’s LIBs still use copper (negative electrode) and aluminum (positive electrode) as current collectors [110,111,112,113] and LiPF6 based non-aqueous aprotic organic solvent electrolytes [114,115,116].

Development of the specific energy and energy density of LIBs (cell level) from 1991 to 2017, following their commercialization by Sony in 1991. Values are based on the standard cylindrical 18650 lithium ion cell and are taken from cell producers’ data sheets and the following references: for values up to 2005 see refs. [3, 11, 28, 117, 118]; 2010: Panasonic-NCR18650A cell; 2012: Panasonic-NCR18650B cell; 2014: NCR1860BF Panasonic-cell; 2015: Panasonic NCR18650GA cell
As an example for the various technological achievements of the numerous companies, the early development stages at Sony are reported. Sony could improve the first generation LIB by changing the anode material from coke (soft carbon) to hard carbon, which offered an enhanced storage capacity, and by using a higher charging voltage (4.2 V instead of 4.1 V). Thus, the second-generation LIB offered an improved specific energy and energy density (120 Wh kg−1; 295 Wh L−1) [117]. The third-generation of Sony’s LIB was further improved by using a graphitic carbon anode material (mesocarbon microbeads (MCMB)) and by using tailored electrolytes, so that the energy could be further increased (155 Wh kg−1; 400 Wh L−1) [117]. By further developments, both the specific energy (80➔ 256 Wh kg−1) and the energy density on cell level (200➔ 697 Wh L−1) could be more than tripled since market introduction until today (Fig. 3).
This incremental performance increase is driven not only by technical achievements, but also by various demands of the consumer and the society such as rising application requirements as well as a growing trend for sustainability and lower costs. Nowadays, the lithium ion technology can be considered as mature, while the run for further performance improvements, i.e. increase of the energy content, dominates the development. Furthermore, the present market for lithium ion batteries seems to be much more complicated than two decades ago, where it was mainly dominated by the requirements for the 3C market (computers, consumer electronics and cameras). Today, there are many additional markets for small-scale devices (lighting, toys, medical devices), for portable power tools, e-bikes, as well as for large-scale applications such as electromobility and stationary energy storage [15, 16, 28]. Particularly in the last decade, there was a rapid increase in the demand for LIBs, as shown by the lithium ion cell sales in Fig. 4.

Lithium ion battery sales worldwide from 2000 to 2016 (cell level); others: power tools, gardening tools, e-bikes, medical devices, etc.; data were obtained from Avicenne [119]
This trend also correlates with the increasing demand from electric-powered vehicles (xEVs), while already ≈42% of the LIB sales (in Wh) account for automobiles in 2016. As there is no indication that the demand for high energy density electrochemical storage devices will stagnate or decline in the near future, the motivation for research and development of novel advanced materials for LIBs is strong, as well as to look at systems beyond LIBs, at the so-called post-lithium ion batteries (PLIBs).
Going for alternatives to lithium ion batteries: development of post-lithium ion technologies
Today, it is widely assumed that the current lithium ion technology will reach its fundamental/intrinsic limits in terms of specific energies and energy densities soon, raising doubts whether this technology is able to cope with the increasing energy storage requirements [30, 33, 120]. In this respect, alternative and advanced battery technologies have been intensively evaluated in recent years, including metal/sulfur, metal/air (or metal/oxygen) systems, batteries based on alternative single (Na+, K+) or multivalent ions (Mg2+, Ca2+), as well as ASSBs. Two of the most promising options in terms of their theoretical energy density are based on lithium metal anodes and have been discussed intensively in the last decade: the Li/O2 and Li/S battery technologies [20,21,22,23,24, 121,122,123,124,125,126].
As a possible power source for electric vehicles, metal/air batteries (such as zinc/air, iron/air, magnesium/air, and lithium/air) have been originally proposed in the 1970s [127]. The interest in this technology was revived in 1996, as Abraham and coworkers introduced the first non-aqueous Li/air battery by using a porous carbon cathode and a carbonate-based polyvinylidene gel polymer electrolyte membrane [128, 129]. In 2006, Ogasawara et al. demonstrated the reversibility for the formation of the discharge product Li2O2 by using MnO2 as catalyst in combination with an organic electrolyte [125], which, in turn, raised the scientific interest for this technology that rapidly got prominent in the following years. However, the initial excitement for the Li/O2 system, that was strongly encouraged by the high theoretical specific energy compared to LIBs, soon slowed down as it was recognized that this technology is quite complex and suffers from various drawbacks such as poor rate capability, electrolyte decomposition during charge as well as by reactive intermediate products such as the lithium superoxide LiO2, high lithium metal anode reactivity and corresponding safety issues as well as high overvoltages, especially during charge. Therefore, it is generally supposed that the Li/O2 technology may not be realizable in the near future as it requires fundamentally new components for the electrolyte or for the separator/membrane [31, 33, 124, 130].
Danuta and Juliusz firstly reported sulfur as positive electrode materials for “electric dry cells and storage batteries” in 1962 [131]. The concept of the Li-sulfur technology was first proposed and patented by Mallory & Co. Inc. in 1966 [132], while the first electrochemical results were reported by Rauh et al. in the late 1970s [133]. However, intensive research and development on rechargeable Li/S batteries started in the 2000s, as can be seen by the exponentially increasing number of publications [24, 126, 134,135,136]. Recently, Sion Power Corporation and Oxis Energy Ltd. both have announced to launch the commercial production of Li/S cells. In 2008, Sion Power already published data for prototype Li/S cells having a specific energy of 350 Wh kg−1 and an energy density of 320 Wh L−1 with a lifetime of ≈50 cycles. In 2016, they reported an even enhanced specific energy and energy density of 400 Wh kg−1 and 800 Wh L−1 with a lifetime of ≈150 cycles [137]. Even though Li/S batteries still suffer from a relatively low cycle life and poor rate performance, it is generally assumed that they will have benefits compared to LIBs for applications where a high specific energy is necessary (e.g. aviation) and limited cycle life can be tolerated (e.g. military applications). Nevertheless, an ongoing improvement in terms of cycle life and other performance parameters is also expected in the next years [20, 23, 130].
Besides the above mentioned lithium-based air and sulfur batteries, a variety of novel battery concepts and technologies approached and developed in the recent years, so that the situation for electrochemical energy storage concepts became much more complex since the commercial introduction of LIBs in the 1990s (Fig. 5). These advanced or alternative post-lithium ion technologies include for example sodium-ion batteries [138,139,140], ASSBs [28, 141, 142], dual-ion or dual-carbon batteries [51,52,53, 56], redox flow batteries [143], magnesium batteries [25, 26, 144, 145], potassium-ion batteries [146, 147], and calcium (-ion) batteries [25, 148, 149]. As all of these technologies are relatively “young” and cannot be considered as mature, large efforts in research and development are needed to improve these novel storage concepts to be able compete with the state-of-the-art lithium ion technology for certain applications. Development progress of a certain technology in the future will be promoted by the increasing demand for higher specific energy and energy density and the request for low costs.

Battery cell chemistries which have been or are considered in R&D to have technological relevance: a up to the 1990s, b today and tomorrow
As all of these advanced post-lithium ion technologies are not established yet, there is also no clear “benchmark” system available and a comparison of these technologies regarding their electrochemical performance seems difficult up to now. Considering the lithium-based systems, in particular the Li/O2 and the Li/S technologies, a broad variety of publications is available that often report a large variation in performance parameters such as discharge capacities, capacity retention and, thus, practically available specific energies and energy densities. This is due to the fact that various types of cell set-ups and components are used and important parameters (e.g., the electrode mass, the amount of the electrolyte with respect to geometrical electrode surface, etc.) are in many cases not stated or stated incompletely, which in turn may put the results in a better light.
Overall, a realistic estimation and comparison of the specific energy and energy density and other performance parameters of different post-lithium ion technologies to state-of-the-art LIBs will only be possible if consistent and standardized cell set-ups and measurement protocols are used and if the authors consistently publish all important parameters that allow a fair comparison.
Approaching the limits: maximizing specific energies and energy densities
Energy density improvements along the battery value chain
For the current evaluation and comparison of the specific energy and energy density for different battery technologies, it is very important to consider the various “levels” along the battery value chain (Fig. 6). The value chain starts at the active material level where—for chemistries where the electrolyte is not actively involved in the charge/discharge reaction—only the anode and cathode active materials are considered, while for a typical lithium ion cell the cathode acts as the lithium ion source and, thus, determines the available lithium capacity after assembly. The common strategy to optimize the energy density on the material level is to improve the materials’ specific and volumetric capacities as well as their operating potentials, i.e., cathode materials should operate at high electrode potentials and anode materials should operate at low electrode potentials in order to maximize the cell voltage.

Schematic illustration of the battery value chain from the material level via the battery cell to the battery system level. In each step, inactive components are added which decrease the practical specific energy/energy density
In the following steps along the battery value chain, the energy density will be decreased by the addition of inactive materials and components. It is widely known that the weight and volume of these inactive components have to be reduced to a minimum for a maximum energy output, however, certain inactive materials or minimum amounts of them are mandatory to ensure the key requirements in terms of performance and safety. [18].
On the electrode level, inactive materials including the polymeric binder [150, 151], the conductive carbon additive [152,153,154] and the current collector [110,111,112,113] have to be added, while the amount and the type of these inactive materials have to be chosen and tailored with respect to the active material properties (e.g., particle size, specific surface area, electronic conductivity, volume expansion during lithiation/de-lithiation, etc.). Furthermore, it is often neglected that the electrode porosity as well as the electrolyte inside the pores have to be considered, as these parameters will have an impact on both the specific energies and energy densities. Major strategies to enhance the energy density on the electrode level include: (1) the optimization of the electrode composition (higher active material/inactive material ratio) in dependence of the preparation technique and parameters [155,156,157,158,159], (2) the increase of electrode mass loading (coating thickness) [160, 161], and (3) decrease of the electrode porosity by calendaring [161,162,163,164].
Many energy density projections reported in literature do either not state to which “level” their calculations refer or do not supply sufficient information, e.g., about the inactive components and their amounts, about the electrode capacity balancing, etc. Furthermore, as also recently reported by Berg et al. [33], evaluations of active materials are often just simple comparisons of their energy density data based on their specific or volumetric capacities and their operating potential (= material level) which, in turn, may lead to misconceptions. In particular, differences in the reaction mechanism (e.g., intercalation/insertion vs. alloying vs. conversion reactions), which are most likely associated with completely different electrode requirements to ensure a suitable electrochemical performance, are neglected in these rather simple energy density projections. One very famous example is the comparison between graphite and silicon-based anode materials for LIBs: While graphite has a high electronic conductivity and only a moderate volume expansion of about ≈10% during lithium uptake via intercalation, silicon displays a poor electronic conductivity and a volume expansion between 300 and 400%, in dependence of the state of lithiation, via an alloying reaction [33, 87, 165]. In turn, a common strategy is to change the binder and use more flexible polymers for Si-based electrodes [166,167,168], which may also interact with the silicon particle surface. Furthermore, the amount of both, the binder and conductive carbon, needs to be enhanced to ensure a high mechanical stability and electronic conductivity of the composite electrode. Currently, state-of-the-art graphite anodes have a high amount of active material in the range of ≈96 wt% with just ≈1 wt% of conductive additive and ≈3 wt% of binder [18]. In contrast, silicon-based anodes, i.e., Si-C composites electrodes (1:3, 1100 mAh g−1), are considered to have a significantly decreased active ratio (≈88 wt%) with increasing amounts inactive components, i.e., ≈2 wt% of conductive additive and ≈10 wt% of binder [18]. Overall, in order to establish more realistic energy density projections, one can only agree with Berg et al. [33] that energy density projections should at least consider the electrode level, but even better the cell or battery pack/system level. Various advanced models for energy calculations at the cell and/or pack level have been proposed in the literature [29, 31, 33, 169].
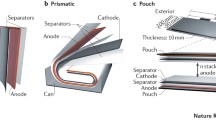
In the next step, by going from the electrode to the cell level (Fig. 6), further inactive materials and parameters, including the separator, the electrolyte (in particular when considering solid electrolytes (SEs)), the current collector tabs, the anode-cathode capacity balancing as well as different components for the cell housing have to be considered for estimation of the energy content. In particular, the cell format does have a major impact on the specific energies and energy densities. Nowadays, three main formats are used for commercial LIB applications: prismatic, cylindrical, and pouch cells. These cells differ in their volume occupancy inside the cell and in the packing density in the battery module and battery pack/system. While cylindrical cells typically exhibit a higher volume utilization on the cell level, prismatic, or pouch cells are more convenient to achieve a high packing efficiency for the battery module [18], Among these cell types, the 18650 cylindrical cell, i.e., a cylindrical cell with a diameter of 18 mm and a height of 65 mm, has the highest production volume with about 660 million cells manufactured annually [170]. Since its market introduction, the energy density of the 18650 cell could be remarkably increased (Fig. 3), which is to a large extent a result of an enhanced active material ratio: While the 18650 cell introduced by Sony in 1991 had an active material weight ratio (anode and cathode) of ≈47%, current cells exhibit an active material weight ratio of ≈61% (Panasonic NCR18650B) [126]. Nevertheless, also other cylindrical cell formats such as 20700 (20 mm diameter, 70 mm height) or 21700 cells (21 mm diameter, 70 mm height) are currently in discussion to replace the 18650 cell for certain applications, as the active material content and, thus, the energy density can be further increased.
On the module level, the single cells are connected in series and are packed in a module container. The module also contains a thermal management system, which is required to regulate the temperature and to keep it in the desired operating range. Even though the use of air as a heat transfer medium is less complicated for technical design, it is also less effective. Therefore, for automotive applications, a thermal management based on liquid coolants such as ethylene glycol or high dielectric constant, non-flammable thermal management fluids (e.g., 3M Novec™ [171]) are commonly used. The modules are also equipped with electronic module control units (MCUs), which have to control the various relevant parameters such as the temperature, the cell voltage, and the current in order to actively prevent failure modes. [170, 172] In the last step, by going to the battery system level (Fig. 6), the battery modules are connected in series and in parallel, to build a battery pack or battery system with a nominal voltage that typically ranges between 300 and 350 V. The battery management system (BMS) consists of the battery control unit (BCU) which collects the data from the single MCUs and develops strategies to protect the cells from their operation outside the threshold conditions, i.e., to prevent overcharging, overdischarging, too high or too low temperatures, short-circuiting, or other failure modes. In order to improve the specific energies and energy densities on the module and pack level, research and development focuses on the investigation of advanced light-weight container or packing materials as well as on optimized strategies for higher packing efficiencies of the cells [170].
Active material contributions to energy density optimization
The total cell capacity a of a lithium ion cell Q cell in [mAh g−1] can be calculated according to the following Eq. [173]:
with Q neg and Q pos being the practical specific discharge capacities of the corresponding anode and cathode material and m inact corresponding to the mass of the inactive components per capacity [g Ah−1]. m inact has been determined to be 6.20 g Ah−1 for a state-of-the-art 18650 cell (Panasonic NCR18650B, 3.35 Ah, 47.5 g), which has a specific energy of 243 Wh kg−1 and an energy density of 676 Wh L−1 [126]. Figure 7 shows the effect of the anode capacity or cathode capacity on the total cell capacity Q cell. As it can be seen in Fig. 7a, Q cell rapidly runs into a plateau area for specific anode capacities beyond 1000 mAh g−1, when the cathode capacity is 200 mAh g−1 (e.g., NCA) or less. From this point of view, a further increase of the anode capacity much beyond a value of 1000 mAh g−1 has only little effect on the overall specific energy of the resulting cell. Only if a cathode with a substantially higher specific capacity would be deployed, a higher capacity anode such as a silicon/carbon composite (Si-C; Q neg = 1000 mAh g−1) [174] or metallic lithium (Li, Q neg = 3862 mAh g−1) [175] would generate a gain in specific energy. In the case of a cathode with a specific capacity of 300 mAh g−1 (e.g., a Li-rich Ni-, Mn-, and Co-based layered oxide; LRNMC) [176], the overall cell capacity may be significantly increased when a high-capacity anode such as Si-C or metallic lithium is used.

Total cell capacity Q cell as a function of a varying specific anode capacities for three different positive electrode materials and b varying specific cathode capacities for three different negative electrode materials. c, d show the plots of the corresponding total cell specific energies E cell (vs. virtual anode and cathodes: c vs. graphite at 0.1 V vs. Li/Li+ d) vs. NMC at 3.8 V vs. Li/Li+), which take the redox potential of the active materials into account. All graphs were calculated with m inact = 6.20 g Ah−1
Figure 7b shows that an increase of the cathode capacity would cause a much more pronounced overall gain for Q cell, as the curves progress much steeper and do not run into a plateau. This clearly illustrates that a high-capacity cathode with a suitable discharge potential (>3 V vs. Li/Li+) would have an immense positive impact on the overall energy content of a LIB [18]. Current high-capacity positive electrode materials are still limited to specific capacities of up to 200 mAh g−1 for NCA or Ni-rich layered oxides such as LiNi0.8Mn0.1Co0.1O2 (NMC-811) [177] or even up to 300 mAh g−1 for Li-rich layered oxides [176, 178, 179], whereas the latter still lack of a suitable long-term cycling stability. In summary, the optimum cell capacity can be achieved, when anodes and cathodes with very high, but also comparably high capacities are deployed.
Fig. 7c, d shows the corresponding total cell specific energy E cell in [Wh kg−1], which can be calculated according to:
where U cell is the nominal voltage of the cell.
These plots take the resulting redox potential of the used active material into account, which have an impact on the resulting specific energy of the cell. For Fig. 7c, a state-of-the-art graphite anode is assumed as a virtual negative electrode (U(graphite) = 0.1 V vs. Li/Li+). While the mean discharge potential of the layered oxides LCO or NCA does not differ significantly, it should be noted that Li-rich oxides on average show a lower discharge potential (~3.4 vs. Li/Li+). Therefore, the advantage of LRNMC in specific energy specific energy [Wh/kg] is lower than is to be expected from the comparison of capacities only.
The same considerations are true, when C-rates, reflecting the charge/discharge currents, and charge/discharge powers, reflecting both charge/discharge currents and charge/discharge voltages, are compared.
Figure 7d shows E cell for different anode materials versus a virtual NMC cathode (U(NMC) = 3.8 V vs. Li/Li+). In this plot, the advantage of the light-weight and low-potential lithium metal over intermetallic anodes such as Si-C composites in terms of specific energy becomes apparent. It should be noted that for simplification all graphs have been calculated with a constant value for m inact of 6.20 g Ah−1. In practical cells this value may vary.
A cell comprising of a NCA cathode with 200 mAh g−1 versus a graphitic anode would yield a specific cell energy of ≈265 Wh kg−1, which is already today almost reached by the Panasonic NCR18650B cell. A change of the anode chemistry from a high density graphite to Si-C (1000 mAh/g, Si:C ratio of 20:80) [173] or Li metal would result in higher specific energies of 279 or 331 Wh kg−1, respectively.
As LIBs are nowadays regularly used in mobile applications, such as portable electronics, and recently also electric vehicles, i.e., applications where the installation space is limited, the battery volume and, thus, its energy density [Wh/L] is generally rated of even higher importance than its mass, i.e., specific energy [Wh/kg]. As battery cells should ideally contain a maximum amount of active material, their volumetric capacities Q vol [mAh cm−3] are just as important as their specific capacities, though they are rarely reported in literature. The volumetric capacity can be simply calculated by multiplication of the practical specific discharge capacity Q grav and the crystallographic density or bulk density ρ of a battery material. For the Si-C composites described here, a specific capacity of Q grav = 1000 mAh g−1 and a bulk density of ρ = 2.2 g cm−3 were assumed.
Q vol[mAh cm −3] = Q grav ∗ ρ
Figure 8 gives an overview of the volumetric capacities for the most commonly discussed, established and upcoming, positive, and negative electrode materials. Despite their very high discharge capacities of up to 300 mAh g−1, Li-rich layered oxides (LRNMC) have a slightly lower crystallographic density of ~4.25 g cm−3 vs. the classical layered oxides such as NMC or NCA (ρ = 4.7 g cm−3). Despite of this, LRNMC oxides provide the highest volumetric capacities of up to 1250 mAh cm−3 among all >3 V cathode materials.

In analogy to the specific capacities and specific energies discussed above, the following two equations describe the dependency of the volumetric capacity Q cell [Ah L−1] and energy density E cell [Wh L−1] from the practical capacities and redox potentials of the used active materials:
\( {Q}_{\mathrm{cell}}\left[\mathrm{mAh}\kern0.62em {\mathrm{L}}^{\hbox{-} 1}\right]=\frac{1}{\frac{1}{Q_{\mathrm{neg}}}+\frac{1}{Q_{\mathrm{pos}}}+{V}_{\mathrm{inact}.}} \)
\( {E}_{\mathrm{cell}}\left[\mathrm{Wh}\kern0.62em {\mathrm{L}}^{\hbox{-} 1}\right]=\frac{U_{\mathrm{cell}}}{\frac{1}{Q_{\mathrm{neg}}}+\frac{1}{Q_{\mathrm{pos}}}+{V}_{\mathrm{inact}.}} \)
with Q pos and Q neg being the practical volumetric discharge capacities [Ah L−1] of the corresponding cathode and anode material, U cell the nominal voltage of the LIB, and V inact the corresponding volume of inactive components per capacity [L Ah−1]. V inact has been determined to be 2.98 mL Ah−1 for a state-of-the-art 18650 cell (Panasonic NCR18650B, 3.35 Ah, 17.6 cm3) and has been used for the calculation of all graphs (Fig. 9) [126]. As in the case of the calculated specific cell energies, the value of V inact may vary for practical LIBs based on different cell chemistries. The three classes of anode materials discussed herein, namely graphite, Si-C composites and lithium metal, provide volumetric capacities of 820, 1270, and 2050 mAh cm−3, respectively [175, 181]. Despite its very high specific capacity, the low-bulk density of metallic lithium (0.53 g cm−3), which is herein calculated to be even slightly lower than that of the Si-C composite, negatively impacts its volumetric capacity. This trivial conclusion on Li metal is noteworthy as the low density of metallic Li is frequently claimed as an advantage for the metallic Li anode in research papers. It is conceivable that a Si-C composite anode in a real cell would likely to require additional void volume for the lithiated silicon to expand into or significant internal stress in the electrode and cell will be generated, as proposed in the models in [182], [183].

Total cell capacity Q cell as a function of a varying volumetric anode capacities for three different positive electrode materials and b varying volumetric cathode capacities for three different negative electrode materials. c, d show the plots of the corresponding total cell energy density E cell, which take the redox potentials of the active materials into account. All graphs were calculated with V inact = 2.98 mL Ah−1
The volumetric cell capacities Q cell as a function of the volumetric anode capacity, depicted in Fig. 9a, show the same trend as in the case of the specific capacities (Fig. 7a). As the bulk densities of the layered oxides show little variation, both Q grav and Q vol increase in the order: LCO < NCA < LRNMC [18]. The analogous graph for Q cell as a function of volumetric cathode capacity is shown in Fig. 9b. As seen before, the Li-metal based cell shows the highest value for Q cell and has a clear advantage of over the cells involving the state-of-the-art graphite anode and Si-C composites.
Figure 9c, d shows plots of the total cell energy density E cell as a function of varying anode and cathode capacities. Again, the nominal cell voltage U cell has been calculated using redox potential of the respective active material versus the potential of the current state-of-the-art counter electrode (NMC or graphite). Due to the higher operating potential and higher bulk density of the layered oxide NCA, the energy density E cell of a NCA-based cell has a slight advantage over a cell based on LRNMC, despite the significantly higher specific and volumetric capacities of LRNMC cathodes. In combination with a high-capacity anode with suitable operating potential, energy densities beyond 700 Wh/L may be realized, which are already almost realized in Panasonic’s NCR18650GA cell with its reported 693 Wh/L. Regarding the impact of the anode chemistry on the total cell energy density, metallic lithium holds the greatest potential among the three compared anode materials in maximizing E cell, due to its immense specific capacity and still very high volumetric capacity (despite its low density) and the lowest electrode potential among all elements (−3.04 V vs. SHE). When a V inact = 2.98 mL Ah−1 is kept constant and a Si-C composite with the chosen properties (Q vol = 1270 mAh cm−3, E = 0.4 V vs. Li/Li+) is used, the energy density of the resulting cell is on par with a graphite-based cell (see Fig. 9d).
Figure 10 shows the schematic representation of various LIB electrode set-ups, for which energy densities were calculated based on an electrode stack energy model [33]. Typical cell parameters of a state-of-the-art LIB such as an electrode porosities of 30% and thicknesses of separator (20 μm) and current collectors (Al = 20 μm, Cu = 10 μm) were selected. A negative to positive capacity balancing (N/P ratio) of 1.1 (=10% excess anode capacity) was chosen for all configurations. Even in the case of lithium metal this value was chosen, although the cell could be in principle be run lithium metal-free, as the cathode is the source of lithium ions [184]. Nevertheless, a continuous immobilization of Li+ is to be expected during cycling, therefore, this excess of Li is chosen.

Schematic representation of various LIB electrode stacks with calculated specific energies and energy densities; from left to right: a C/NCA, b Li/NCA, c Si-C/NCA, and d a thick Li/NCA configuration. The shown scale arrows refer to the thickness of the composite cathode of 100 μm for the first three stacks a–c and 165 μm for the stack d, which is as thick as stack a
The calculations of stacks a–c (Fig. 10) are based on a NCA cathode with a high electrode thickness of 100 μm. The resulting cell specific energies and energy densities of the C/NCA, Li/NCA, and Si-C/NCA electrode stacks amount to 296 Wh/L/765 Wh/L, 435 Wh/kg/1177 Wh/L, and 402 Wh/kg/1213 Wh/L, respectively. The substitution of today’s graphitic negative electrode by Li metal would result in a ~50% gain in specific energy and a ~55% gain in energy density. Among the three thin cell set-ups, the Si-C/NCA stack exhibits the highest energy density of 1213 Wh/L, although a higher electrode porosity of 50% (instead of 30%) for the Si-C anode has been taken into account in this energy density calculation.
The positive impact of high-capacity anode materials on the overall energy density becomes even more obvious, when the same volume is utilized by the energy-optimized LIB stack, e.g., in an 18650-type cell. This is exemplified in Fig. 10d, where a significantly thicker NCA electrode of 165 μm is used instead of a 100 μm electrode. Leaving aside the practical feasibility of such a thick electrode due to diffusion limitations within the composite cathode, the resulting stack energies of 474 Wh/kg/1286 Wh/L are even higher than for the thinner Li/NCA stack shown in Fig. 10b.
Fig. 11 shows a comparison of the currently well-established lithium ion technology with the currently most discussed post-lithium ion technologies such as lithium/sulfur (Li/S) or lithium/oxygen (Li/O2) batteries on the cell level in an energy density vs. specific energy diagram (Wh L−1 = Wh kg−1).

Energy density vs. specific energy plot of today’s LIBs (dark blue) in comparison to energy-optimized LIBs (light blue), classical Li-metal batteries (CLIMs; green) and post-lithium ion technologies such as lithium/sulfur (Li/S) as well as lithium/oxygen batteries (Li/O2) on the cell level. The dotted gray line represents the parity of energy density and specific energy (Wh/L = Wh/kg). Values are partly taken from reference [186]
The dark blue ellipse represents the range of today’s LIBs, which can significantly vary depending on the chosen cell design and field of application (high-power or high-energy cell). In this graph, a major advantage of LIBs becomes obvious, namely its very high energy density, which is usually 2–3 times higher than the specific energy. Thus, the dark blue ellipse lies clearly above the gray line of specific energy = energy density parity. Through the implementation of novel active materials with even higher volumetric and specific capacities (Si-based anodes, Li-rich layered (LRNMC) cathodes), the energy contents of current LIBs may be further increased. These “energy-optimized” LIBs are represented by the light-blue ellipse in Fig. 11, with specific energies/energy densities of ≈400 Wh/kg/800 Wh/L.
If the technical challenges of the metallic lithium anode, especially regarding safety and processing, could be overcome in the near future, this would pave the way to batteries with even higher energy contents. The most intensely discussed post-lithium ion technologies Li/S and Li/O2 (yellow and red ellipses) both require a lithium metal negative electrode. Beside their pronounced technical challenges for realization, their energy densities are not likely to be much higher than that for the specific energy, which is visualized by their ellipses laying on the dotted gray parity line [31, 126, 185]. A lithium metal anode could, however, also been combined with existing or upcoming high-energy density/specific energy cathode materials. As this cell set-up would involve a lithium metal anode, it should no longer be termed as lithium ion cell. We refer to these cells as classical lithium metal batteries (CLIMs), which are represented by the green ellipse in Fig. 11. In an optimistic scenario, CLIMs may reach specific energies of up to 500 Wh/kg and possibly exceed energy densities of 1000 Wh/L.
Current LIBs therefore hold a clear advantage when it comes to energy density, which is of high importance for the fast-growing market of mobile consumer applications and electromobility. The implementation of new high-capacity positive electrode materials and the potential renaissance of the lithium metal negative electrode promises even higher energies. In the future, practical Li/S or even Li/O2 battery cells are likely to show advantages in terms of energy per weight over LIBs and should, therefore, be of interest for more mass-critical fields of application such as aviation.
From lithium ion cells back to lithium metal systems: the all-solid-state battery concept
After first developments of solid electrolytes (SEs) for rechargeable lithium metal solid state batteries in the early 1980s [187], the concept of an all-solid-state battery (ASSB) remained less economically attractive with a few exceptions (e.g., the lithium/iodine battery [188]) due to performance drawbacks and safety issues regarding to the lithium metal anode [36]. The discovery of new ceramic electrolytes with sufficient conductivity at room temperature in recent years led to a renewing interest in establishing a performance-competitive ASSB. SEs can be promising alternatives to liquid electrolytes as their solid nature can overcome several disadvantages of organic solvent-based LIBs. Compared to current liquid organic electrolytes in LIBs, solid ceramic (inorganic) electrolytes can provide safety improvements as they are not flammable (though they can be oxidized), avoid leakages and gas formation from/in the cell, and are stable against high temperatures [27]. LIBs with SE show further only minor self-discharge due to a negligible electronic conductivity of the SE. [189] New inorganic electrolytes (e.g., Li7P3S11) [190], which are single-ion conductors (Li+ only conduction) with high-ionic conductivities (also at room temperature), were reported to enable higher charging rates and quicker charging times compared to liquid electrolytes. At high charging rates, high charge concentrations can occur in liquid electrolyte solvents which can be avoided in SEs. Moreover, SEs can enable new cell chemistries and cell designs [191].Unlike liquid electrolytes, SEs do not require a costly separator.
On the one hand, SEs show the advantage to hinder the exchange of redox-active species that could shuttle between the positive and negative electrode which may often lead to a capacity loss by internal reactions. Therefore, SEs may be able to overcome common problems like the diffusion of soluble polysulfides in lithium/sulfur cells (“polysulfide shuttle”) [122] or a leaching of transition metal ions from LIB cathode materials [192,193,194,195] or metal cations from Li storage anodes [196]. The mechanical rigidity of ceramic electrolytes, on the other hand, may suppress the formation of lithium dendrites allowing the usage of lithium metal as anode material [197]. Furthermore, novel cell designs can be achieved in ASSBs that reduce the amount of inactive parts (cell housing, wiring, etc.) on the battery system/pack level. Such a concept represents a bipolar stacking of anodes and cathodes with SE separation (see Fig. 12, right) that leads to smaller lateral dimensions of the cell as well as to higher inherent cell voltages. Compared to a series connection of conventional lithium ion cells (Fig. 12, left), the bipolar stack can offer a higher specific energy on the system level due to less inactive parts and better use of space is possible [198,199,200].

Comparison of two cell designs: series connection of conventional lithium ion cells (left). Bipolar stacking of anodes and cathodes separated by SE layers in an ASSB (right). Adopted from [199]
In general, SEs for ASSBs can be divided into two material classes. The first class comprises of organic solid polymers, known for a few decades as ionic conductors in lithium metal and lithium ion cells [187, 201,202,203,204]. Due to their usually malleable and elastic nature, just like electrode binders, polymers could be the ideal SE to compensate large volume changes of the active materials in the electrodes. However, presently the ionic conductivities of polymer electrolytes are too low at room temperature for operation in high-power/high-energy density batteries like in automotive applications. Only at elevated temperatures (e.g., 80 °C), polymer-based ASSBs can be competitive with conventional LIBs, although also the rate capability of these high-temperature systems is limited [28]. Further developments concerning charging rate improvements and higher energy densities will be the main challenge for the next generation of polymeric electrolytes. Also the use of jellified polymers or polymer/liquid hybrids may be an option [205,206,207,208].
The second class covers inorganic crystalline or glass-ceramic materials. In the past, many research groups aimed to improve the ionic conductivity of oxide- and phosphate-based ceramics, i.e., of the LISICON-type lithium ion conductor (lithium super ion conductor, Li14ZnGe4O16) [209], the LATP lithium ion conductor (Li1+x Al x Ti2−x (PO4)3) with NASICON-type structure (sodium super ion conductor) [210], perovskite (La0.5Li0.5TiO3) [211], garnet (Li7La3Zr2O12, LLZO) [212], and “lithium phosphorus oxide nitride” (Li2.88PO3.73N0.14, LiPON) classes [213]. Although these materials exhibit ionic conductivities in the order of 10−3 to 1 mS cm−1 [214], major drawbacks are their rigidity, inflexibility, and brittleness which makes the electrode processing challenging. Here, sulfide glasses or glass-ceramics with comparable lithium ion conductivities may be more suitable due to their higher ductility [215]. However, sulfide glasses are unstable against moisture and tend to decompose under formation of H2S [216]. Recent progress on sulfide-based structures led to lithium ion conductors that exhibit room temperature conductivities comparable to those of liquid electrolytes. Record conductivities are shown for Li7P3S11 (LPS), a meta-stable crystalline phase in the Li2S-P2S5 system, and Li10GeP2S12 of the thio-LISICON class with 17 and 12 mS cm−1, respectively [190, 217]. Most promising novel lithium ion conducting materials, however, are less thermodynamically stable and form a passivating interphase on lithium metal surfaces [218, 219]. Some materials can further easily be oxidized at elevated potentials on the cathode side similar to liquid carbonate-based electrolytes [220]. Therefore, protective coatings on the SE particles are necessary to overcome the high surface reactivity of reactive compounds.
A closer look at the electrode and cell design of SE-based cells (see Fig. 13) reveals the main challenges of an ASSB cell processing.

Schematic illustrations of a conventional lithium ion cell containing a liquid electrolyte and a polypropylene (PP) separator (left) and ASSB cell with an electrolyte and separating layer consisting of an ionically conductive material (right). See also [28]
While liquid electrolytes can easily impregnate the porous structure of composite electrodes and the separator, resulting in an optimized contact between electrolyte and active material, in an ASSB cell the SE has to be premixed with the electrode materials to obtain a mutual penetration (“entanglement”) of the active material and the SE for a high contact area and optimal ionic conductivity. To ensure a high cycle life, the SE and electrode material need to stay in contact and, thus, a fixation of a stable interphase between both compounds is mandatory. The tasks for the ASSB cell production are firstly to guarantee a homogeneous distribution of the SE particles in the electrode for an optimal entanglement of both substances. Secondly, a subsequent sintering step after the assembly of anode, separating and cathode layer might be necessary to establish a stable contact between all particles. To maintain the close contact during operation, a compressing cell housing with an external pressure on the cell stack should be considered [28]. In a third place, strategies have to be developed for a high-speed production of ASSB cells that are economically competitive with current LIB cell production techniques.
Beside safety reasons and other benefits (see above), SEs are often associated with a remarkable increase potential for the specific energy (Wh kg−1) in ASSBs compared to conventional organic solvent-based LIBs. However, when the electrolyte densities of these systems are compared (see Fig. 14), it is obvious that SEs can exhibit up to nearly five times higher values than carbonate solvent-based liquid electrolytes.

Densities of selected liquid (blue) and solid (yellow) electrolytes for lithium ion cells. LPS: Li7P3S11, LATP: Li1.5Al0.5Ti1.5(PO4)3; garnet: Li7La3Zr2O12
Hence, the mass of an ASSB cell with identical cell parameters such as electrolyte content and electrode thicknesses is inevitably higher resulting in a lower specific energy [Wh/kg]. For example, specific energy calculations of electrode stacks with parameters listed in Table 2, containing the active materials graphite and a Ni-rich layered oxide cathode LiNi0.8Mn0.1Co0.1O2 (NMC-811) as depicted in Fig. 13, deliver values of 302 Wh kg−1 for cells with carbonate-based electrolyte and 278 Wh kg−1 for an ASSB cell employing the light SE LPS. For this calculation, the weight of the cell housing material was neglected.
Even though the rather light SE Li7P3S11 (LPS) was considered for the calculation, the conventional carbonate-based LIB cell excels an ASSB cell in terms of specific energy, if a comparable cell set-up with the state-of-the-art active materials is assumed.
In order to establish a high-energy density and high-specific energy ASSB cell, the most promising step seems to switch from the lithium ion technology back to lithium metal-based cells (see Fig. 15) [197].

Schematic comparison of ASSB cells with graphite (left) and lithium metal (right) as anode materials. See also [28]
Battery-grade lithium metal has never been a cheap material, mostly because of the costs of processing to thin foil or particles. Moreover, raw material prices do fluctuate strongly in general and especially the lithium metal price has increased steeply recently [221] and is expected to increase further. This will have an effect not only on ASSBs, but also on other Li metal chemistries, such as Li/S, Li/air, and CLIM (see above), making Li metal anode chemistries presently (2017) not cost-competitive to lithium ion cell chemistries [222].
Nevertheless, Li metal is finding strong attention in the community, as anode material due to its high theoretical discharge capacity of 3860 mAh g−1, which reduces the overall cell weight as well as the cell dimensions and improves therefore the energy densities and specific energies compared to graphite-based and most other lithium ion storage anode material-based cell chemistries. Calculations of a model cell stack with the parameters of Table 2 containing a 30 μm thick lithium metal anode instead of the 120 μm graphite layer show a specific energy of 426 Wh kg−1 for a LPS-based or even 324 Wh kg−1 for a garnet-based ASSB. Lithium metal-based ASSB cells exhibit, thus, a remarkable specific energy increase of 53% compared to its graphite-based version. This demonstrates the enormous potential of lithium metal anodes that seem to be only applicable with solid inorganic electrolytes to date due to their potential (but not so far sufficiently proven) ability to prevent the formation of hazardous lithium dendrites occurring in liquid organic solvent-based electrolytes [223, 224]. Recent developments of cathode materials with thin SE coatings [225, 226] reveal another option to raise further the specific energy of ASSBs. Instead of a mutual penetration of SE and cathode material, thin ionic conductive coatings could fulfill the lithium ion transport while reducing the fraction of SE in the cathode at the same time. The relationship between the SE content within the cathode and the specific energy of a model cell containing a lithium metal anode and a NMC-811 cathode is presented in Fig. 16.

Specific energy calculations of a model ASSB cell stack containing lithium metal (30 μm) and NMC-811 (100 μm) as active materials in relationship to the SE volume fraction in the cathode for SEs with different densities. A separation layer thickness of 20 μm was assumed. For comparison, calculations of a similar model cell stack based on liquid electrolyte with different cathode porosity is depicted as a blue line
The calculations show that the reduction of the SE volume fraction in the cathode from 30% to a more realistic value of 15% lead to a specific energy of 479 Wh kg−1 for the light LPS and 397 Wh kg−1 for the heavier garnet type SE. When high-energy cells are considered, it becomes obvious that the application of a light SE (LPS) is the most realistic scenario for ASSB cells. For a certain specific energy value, e.g., 450 Wh kg−1 (see dashed line in Fig. 16), the calculation reveals that a model cell with the light LPS contains a reasonable SE volume fraction of 25%, whereas a model cell with a heavy garnet SE allows only a SE volume fraction of 5%. In relation to that, a liquid-based model cell with a lithium anode would exhibit a porosity of 35%. It should be noted at this point that research of SE-coated particles leading to a lower SE amount in the cell is still in the very early stages and that the calculations rather target the revealing of possible ways to optimize the specific energy of an ASSB cell.
Our calculations as well as current research trends regarding SEs suggest that the future of ASSBs with high energy density and specific energy relies on the application of lithium metal and an optimized cell processing. Figure 17 summarizes our model cell calculations and shows a possible development path from the liquid-based state-of-the-art LIB technology to the high-energy all-solid-state battery based on lithium metal. In addition, the energy density, which is considered to be important for mobile applications, does not change significantly when a model cell stack is switched from liquid to SEs under the same cell design parameters.

Schematic illustrations and specific energy calculations of model ASSB cells containing nickel manganese cobalt oxide (NMC-811) cathodes, graphite (Gr), or lithium metal (Li) as active materials. A separation layer of 20 μm and 30 vol% of carbonate-based liquid electrolyte or LPS-based (SE) electrolyte in the electrodes were used for the calculation. A single asterisk indicates that a SE content of 15 vol% was considered for the SE-coated NMC-811 cathode particles
Although the calculated values of the SE-based cell systems are quite impressive, the realization of these concepts remains challenging. All the possible ASSB systems offer, in our point of view, at least one major task to solve. In the case of a SE-based cell with lithium ion storage electrodes (graphite/NMC), a high contact area between SE and active material has to be established and conserved during operation. When lithium metal is applied as anode in the second case, new strategies must be found to overcome the instability of the thermodynamically less stable SEs (for example LPS). In the third case, SE-coated cathode particles should be highly ionically as well as electronically conductive. While the first case can be managed with a deeper understanding of the interfacial reactions between SE and active material and a thereon fitted cell design, the second and third cases can be expected to be rather long-term challenges. In all three cases, the complex cell mechanics (electrode/electrolyte contacts) of shape-changing electrodes have to be considered, as lithium ion storage materials do expand and contract during charge and discharge [227], whereas Li metal dissolves and deposits during charge and discharge, thus, creating a dynamic interface between the anode and the SE. Hence, for SE-based cells, zero-strain electrode and cell designs need to be created. At contrast, these mechanics play a minor role in liquid electrolyte cells where the electrolyte is able to conform a “wetted interface” with electrodes of shape-changing electrodes throughout charge/discharge cycling.
Thinking in generations and roadmaps
The history of electrochemical energy storage is more than 200 years old. With LIBs nowadays considered as the state-of-the-art battery technology for various small- and large-scale applications, many primary (= non-rechargeable) and secondary (= rechargeable) battery systems that have been developed before the commercial introduction of LIBs in 1991 can be expressed as “Before-LIB.”.A few of these “old” battery systems still have commercial relevance until today (parallel-to-LIB), including for example the nickel-based systems (Ni-Cd, Ni-MH), the lead-acid battery or various primary lithium metal systems (Fig. 18).

Terminology of battery systems: “Before-lithium ion,” “Parallel-to-lithium ion,” and “Post-lithium ion”
Based on its unique mix of beneficial performance (high specific energy and energy density, long cycle and calendar life, high safety) and cost properties, the LIB has become the benchmark for eventual future battery systems, the post-lithium ion technologies (post-LIBs, PLIBs) including ,e.g., lithium metal-based systems (CLIM, Li/S, Li/O2, ASSB, etc.). Experiences that have been made in the development of LIBs and Before-LIBs so far, will help to further optimize LIBs and develop advanced PLIBs.
For the upcoming years, it is a common sense that research and development will significantly improve the specific energies and energy densities of LIBs. There is, however, agreement in the community that the limits of standard (= known) chemistries will be soon reached on the material level, so that further energy density increases are only possible up to a doubling of the current energy density values. Further improvements concern the inactive cell material components, which are expected to reduce the weight and volume. In addition to better cell chemistries, optimized cell-, module-, and system-components are expected on the engineering/system level. However, currently it seems rather uncertain, whether these optimized LIBs can be surpassed in the future by post-lithium ion technologies, or even post-lithium technologies (containing cell chemistries without Li) and even if so, whether this will lead to removal of LIBs from the market. Overall, from today’s point of view it seems quite clear that as in the past and today, there will not only be one technology for all applications (monopoly), but different battery systems, which can be especially suitable or combined for a particular application (technology diversity, Fig. 19).

Development of battery technologies
References
IEA (2017) https://www.iea.org/ (Accessed January 12, 2017)
Nagaura T (1991) Prog Batteries Solar Cells 10:218
Nishi Y (2001) Lithium ion secondary batteries; past 10 years and the future. J Power Sources 100(1–2):101–106
Tarascon JM, Armand M (2001) Issues and challenges facing rechargeable lithium batteries. Nature 414(6861):359–367
Winter M, Brodd RJ (2004) What are batteries, fuel cells, and supercapacitors? Chem Rev 104(10):4245–4269
Armand M, Tarascon JM (2008) Building better batteries. Nature 451(7179):652–657
Scrosati B, Garche J (2010) Lithium batteries: status, prospects and future. J Power Sources 195(9):2419–2430
Etacheri V, Marom R, Elazari R, Salitra G, Aurbach D (2011) Challenges in the development of advanced Li-ion batteries: a review. Energy Environ Sci 4(9):3243–3262
Scrosati B, Hassoun J, Sun Y-K (2011) Lithium-ion batteries. A look into the future. Energy Environ Sci 4(9):3287–3295
Wagner R, Preschitschek N, Passerini S, Leker J, Winter M (2013) Current research trends and prospects among the various materials and designs used in lithium-based batteries. J Appl Electrochem 43(5):481–496
Crabtree G, Kócs E, Trahey L (2015) The energy-storage frontier: lithium-ion batteries and beyond. MRS Bull 40(12):1067–1078
Larcher D, Tarascon JM (2015) Towards greener and more sustainable batteries for electrical energy storage. Nat Chem 7(1):19–29
Schipper F, Aurbach D (2016) A brief review: past, present and future of lithium ion batteries. Russ J Electrochem 52(12):1095–1121
Deng D (2015) Li-ion batteries: basics, progress, and challenges. Energy Sci Eng 3(5):385–418
Blomgren GE (2017) The development and future of lithium ion batteries. J Electrochem Soc 164(1):A5019–A5025
Tarascon JM (2016) The Li-ion battery: 25 years of exciting and enriching experiences. Electrochem Soc Interface 25(3):79–83
Besenhard JO, Winter M (1998) Insertion reactions in advanced electrochemical energy storage. Pure Appl Chem 70(3):603–608
Andre D, Kim S-J, Lamp P, Lux SF, Maglia F, Paschos O, Stiaszny B (2015) Future generations of cathode materials: an automotive industry perspective. J Mater Chem A 3:6709–6732
Patry G, Romagny A, Martinet S, Froelich D (2014) Cost modeling of lithium-ion battery cells for automotive applications. Energy Sci Eng 3(1):71–82
Bruce PG, Freunberger SA, Hardwick LJ, Tarascon JM (2012) Li-O2 and Li-S batteries with high energy storage. Nat Mater 11(1):19–29
Capsoni D, Bini M, Ferrari S, Quartarone E, Mustarelli P (2012) Recent advances in the development of Li-air batteries. J Power Sources 220:253–263
Christensen J, Albertus P, Sanchez-Carrera RS, Lohmann T, Kozinsky B, Liedtke R, Ahmed J, Kojic A (2012) A critical review of Li/air batteries. J Electrochem Soc 159(2):R1–R30
Bresser D, Passerini S, Scrosati B (2013) Recent progress and remaining challenges in sulfur-based lithium secondary batteries—a review. Chem Commun 49(90):10545–10562
Manthiram A, Fu Y, Chung S-H, Zu C, Su Y-S (2014) Rechargeable lithium–sulfur batteries. Chem Rev 114(23):11751–11787
Canepa P, Sai Gautam G, Hannah DC, Malik R, Liu M, Gallagher KG, Persson KA, Ceder G (2017) Odyssey of multivalent cathode materials: open questions and future challenges. Chem Rev 117(5):4287–4341
Besenhard JO, Winter M (2002) Advances in battery technology: rechargeable magnesium batteries and novel negative-electrode materials for lithium ion batteries. ChemPhysChem 3(2):155–159
Kim JG, Son B, Mukherjee S, Schuppert N, Bates A, Kwon O, Choi MJ, Chung HY, Park S (2015) A review of lithium and non-lithium based solid state batteries. J Power Sources 282:299–322
Janek J, Zeier WG (2016) A solid future for battery development. Nature Energy 1:16141
Nelson P, Gallagher K, Bloom I, Dees D (2011) Modeling the performance and cost of lithium-ion batteries for electric-drive vehicles. Chemical Sciences and Engineering Division. Argonne National Laboratory, Argonne, IL US
Thackeray MM, Wolverton C, Isaacs ED (2012) Electrical energy storage for transportation-approaching the limits of, and going beyond, lithium-ion batteries. Energy Environ Sci 5(7):7854–7863
Gallagher KG, Goebel S, Greszler T, Mathias M, Oelerich W, Eroglu D, Srinivasan V (2014) Quantifying the promise of lithium-air batteries for electric vehicles. Energy Environ Sci 7(5):1555–1563
Van Noorden R (2014) A better battery. Nature 507(7490):26–28
Berg EJ, Villevieille C, Streich D, Trabesinger S, Novák P (2015) Rechargeable batteries: grasping for the limits of chemistry. J Electrochem Soc 162(14):A2468–A2475
Gröger O, Gasteiger HA, Suchsland J-P (2015) Review—electromobility: batteries or fuel cells? J Electrochem Soc 162(14):A2605–A2622
Wood Iii DL, Li J, Daniel C (2015) Prospects for reducing the processing cost of lithium ion batteries. J Power Sources 275:234–242
Scrosati B (2011) History of lithium batteries. J Solid State Electrochem 15(7–8):1623–1630
Placke T, Winter M (2015) Batterien für medizinische Anwendungen. Z Herz- Thorax- Gefäßchir 29(2):139–149
Bieker P, Winter M (2015) Was braucht man für eine Super-Batterie? Chem Unserer Zeit 50(1):26–33
Winter M, Besenhard JO (1999) Wiederaufladbare Batterien. Teil 1: Akkumulatoren mit wäßriger Elektrolytlösung. Chem Unserer Zeit 33(5):252–266
Owens BB (1986) Batteries for implantable biomedical devices. Plenum Press, New York
Rüdorff W, Hofmann U (1938) Über Graphitsalze. Z Anorg Allg Chem 238(1):1
McCullough FP, Beale AF (1989) Electrode for use in secondary electrical energy storage devices—avoids any substantial change in dimension during repeated electrical charge and discharge cycles. US Pat 4:865,931
McCullough FP, Levine A, Snelgrove RV (1989) Secondary battery. US Pat 4:830,938
McCullough FP (1996) Flexible carbon fiber, carbon fiber electrode and secondary energy storage devices. US Pat 5:518,836
McCullough FP (1996) Flexible carbon fiber electrode with low modulus and high electrical conductivity, battery employing the carbon fiber electrode, and method of manufacture. US Pat 5:532,083
Carlin RT, Delong HC, Fuller J, Trulove PC (1994) Dual intercalating molten electrolyte batteries. J Electrochem Soc 141(7):L73–L76
Carlin RT, Fuller J, Kuhn WK, Lysaght MJ, Trulove PC (1996) Electrochemistry of room-temperature chloroaluminate molten salts at graphitic and nongraphitic electrodes. J Appl Electrochem 26(11):1147–1160
Dahn JR, Seel JA (2000) Energy and capacity projections for practical dual-graphite cells. J Electrochem Soc 147(3):899–901
Seel JA, Dahn JR (2000) Electrochemical intercalation of PF6 into graphite. J Electrochem Soc 147(3):892–898
Placke T, Bieker P, Lux SF, Fromm O, Meyer HW, Passerini S, Winter M (2012) Dual-ion cells based on anion intercalation into graphite from ionic liquid-based electrolytes. Z Phys Chem 226:391–407
Placke T, Fromm O, Lux SF, Bieker P, Rothermel S, Meyer HW, Passerini S, Winter M (2012) Reversible intercalation of bis (trifluoromethanesulfonyl) imide anions from an ionic liquid electrolyte into graphite for high performance dual-ion cells. J Electrochem Soc 159(11):A1755–A1765
Rothermel S, Meister P, Schmuelling G, Fromm O, Meyer HW, Nowak S, Winter M, Placke T (2014) Dual-graphite cells based on the reversible intercalation of bis (trifluoromethanesulfonyl) imide anions from an ionic liquid electrolyte. Energy Environ Sci 7(10):3412–3423
Read JA, Cresce AV, Ervin MH, Xu K (2014) Dual-graphite chemistry enabled by a high voltage electrolyte. Energy Environ Sci 7(2):617–620
Zhang X, Tang Y, Zhang F, Lee C-S (2016) A novel aluminum–graphite dual-ion battery. Adv Energy Mater 6(11):1502588–1502593
Tong X, Zhang F, Ji B, Sheng M, Tang Y (2016) Carbon-coated porous aluminum foil anode for high-rate, long-term cycling stability, and high energy density dual-ion batteries. Adv Mater 28(45):9979–9985
Miyoshi S, Nagano H, Fukuda T, Kurihara T, Watanabe M, Ida S, Ishihara T (2016) Dual-carbon battery using high concentration LiPF6 in dimethyl carbonate (DMC) electrolyte. J Electrochem Soc 163(7):A1206–A1213
Meister P, Siozios V, Reiter J, Klamor S, Rothermel S, Fromm O, Meyer HW, Winter M, Placke T (2014) Dual-ion cells based on the electrochemical intercalation of asymmetric fluorosulfonyl-(trifluoromethanesulfonyl) imide anions into graphite. Electrochim Acta 130 (0):625–633
Onagi N, Hibino E, Okada S, Ishihara T (2014) Nonaqueous electrolyte secondary battery. US20140186696 A1
Winter M, Besenhard JO (1999) Wiederaufladbare Batterien. Teil 2: Akkumulatoren mit nichtwäßriger Elektrolytlösung. Chem Unserer Zeit 33(6):320–332
Peled E (1979) The electrochemical-behavior of alkali and alkaline-earth metals in non-aqueous battery systems - the solid electrolyte interphase model. J Electrochem Soc 126(12):2047–2051
Besenhard JO, Winter M, Yang J, Biberacher W (1995) Filming mechanism of lithium-carbon anodes in organic and inorganic electrolytes. J Power Sources 54(2):228–231
Peled E, Golodnitsky D, Ardel G (1997) Advanced model for solid electrolyte interphase electrodes in liquid and polymer electrolytes. J Electrochem Soc 144(8):L208–L210
Winter M, Appel WK, Evers B, Hodal T, Moller KC, Schneider I, Wachtler M, Wagner MR, Wrodnigg GH, Besenhard JO (2001) Studies on the anode/electrolyte interface in lithium ion batteries. Chem Mon 132(4):473–486
Edström K, Herstedt M, Abraham DP (2006) A new look at the solid electrolyte interphase on graphite anodes in Li-ion batteries. J Power Sources 153(2):380–384
Winter M (2009) The solid electrolyte interphase—the most important and the least understood solid electrolyte in rechargeable Li batteries. Z Phys Chem 223(10–11):1395–1406
Verma P, Maire P, Novak P (2010) A review of the features and analyses of the solid electrolyte interphase in Li-ion batteries. Electrochim Acta 55(22):6332–6341
An SJ, Li J, Daniel C, Mohanty D, Nagpure S, Wood III DL (2016) The state of understanding of the lithium-ion-battery graphite solid electrolyte interphase (SEI) and its relationship to formation cycling. Carbon 105:52–76
Schranzhofer H, Bugajski J, Santner H, Korepp C, Möller K-C, Besenhard J, Winter M, Sitte W (2006) Electrochemical impedance spectroscopy study of the SEI formation on graphite and metal electrodes. J Power Sources 153(2):391–395
Root MJ (2013) Medical Device Batteries. In: Brodd RJ (Ed.) Batteries for sustainability—selected entries from the Encyclopedia of Sustainability Science and Technology. Springer, New York
Eichinger G, Semrau G (1990) Lithiumbatterien I. Chemische Grundlagen. Chem Unserer Zeit 24(1):32–36
Eichinger G, Semrau G (1990) Lithiumbatterien II. Entladereaktionen und komplette Zellen. Chem Unserer Zeit 24(2):90–96
Brandt K (1994) Historical development of secondary lithium batteries. Solid State Ionics 69(3–4):173–183
Watanabe K, Fukuda M (1970) Primary cell for electric batteries. US Patent No 3:536,532
Schneider AA, Moser JR (1972) Primary cells and iodine-containing cathodes therefore. US Patent 3:674,562
Julien C, Mauger A, Vijh A, Zaghib K (2016) Lithium batteries. Science and Technology, Springer International Publishing, Switzerland
Reddy TB (2010) Linden’s Handbook of Batteries, 4th Edition. McGraw-Hill Education, New York
Whittingham MS (1976) Electrical energy-storage and intercalation cehmistry. Science 192(4244):1126–1127
Whittingham MS (1978) Chemistry of intercalation compounds—metal guests in chalcogenide hosts. Prog Solid State Chem 12(1):41–99
Whittingham MS (2004) Lithium batteries and cathode materials. Chem Rev 104(10):4271–4301
Pereira N, Amatucci GG, Whittingham MS, Hamlen R (2015) Lithium-titanium disulfide rechargeable cell performance after 35 years of storage. J Power Sources 280:18–22
Fouchard D, Taylor JB (1987) The Molicel rechargeable lithium system—multicell aspects. J Power Sources 21(3–4):195–205
Brandt K, Laman FC (1989) Reproducibility and reliability of rechargeable lithium molybdenum-disulfide batteries. J Power Sources 25(4):265–276
Robillard C (2005) Proc IEEE Power Engineering Society General Meeting. San Francisco, CA, June 12–16:1223–1227
Dan P, Mengeritsky E, Aurbach D, Weissman I, Zinigrad E (1997) More details on the new LiMnO2 rechargeable battery technology developed at Tadiran. J Power Sources 68(2):443–447
Mengeritsky E, Dan P, Weissman I, Zaban A, Aurbach D (1996) Safety and performance of Tadiran TLR-7103 rechargeable batteries. J Electrochem Soc 143(7):2110–2116
Fouchard D, Lechner L (1993) Analysis of safety and reliability in secondary lithium batteries. Electrochim Acta 38(9):1193–1198
Winter M, Besenhard JO, Spahr ME, Novak P (1998) Insertion electrode materials for rechargeable lithium batteries. Adv Mater 10(10):725–763
Heine J, Hilbig P, Qi X, Niehoff P, Winter M, Bieker P (2015) Fluoroethylene carbonate as electrolyte additive in tetraethylene glycol dimethyl ether based electrolytes for application in lithium ion and lithium metal batteries. J Electrochem Soc 162(6):A1094–A1101
Lazzari M, Scrosati B (1980) Cyclable lithium organic electrolyte cell based on 2 intercalation electrodes. J Electrochem Soc 127(3):773–774
Scrosati B (1992) Lithium rocking chair batteries—an old concept. J Electrochem Soc 139(10):2776–2781
Mizushima K, Jones PC, Wiseman PJ, Goodenough JB (1980) LixCoO2—a new cathode material for batteries of high-energy density. Mater Res Bull 15(6):783–789
Winter M, Besenhard JO (1999) Lithiated carbons. In: Besenhard JO (ed) Handbook of Battery Materials. Wiley-VCH Verlag GmbH, Weinheim, pp 383–418
Winter M, Möller K-C, Besenhard JO (2003) Carbonaceous and graphitic anodes. In: Nazri G-A, Pistoia G (eds) Lithium batteries: Science and Technology. Springer US, Boston, pp 145–194
Juza R, Wehle V (1965) Lithium-Graphit-Einlagerungsverbindungen. Naturwissenschaften 52(20):560
Bagouin M, Guerard D, Herold A (1966) Action de la vapeur de lithium sur le graphite. Comptes Rendus Hebdomadaires Des Seances De L Academie Des Sciences Serie C 262(7):557
Guerard D, Herold A (1972) New method for preparation of insertion compounds of lithium in graphite. Comptes Rendus Hebdomadaires Des Seances De L Academie Des Sciences Serie C 275(11):571
Guerard D, Herold A (1975) Intercalation of lithium into graphite and other carbons. Carbon 13(4):337–345
Dey AN, Sullivan BP (1970) Electrochemical decomposition of propylene carbonate on graphite. J Electrochem Soc 117(2):222
Arakawa M, Yamaki JI (1987) The cathodic decomposition of propylene carbonate in lithium batteries. J Electroanal Chem 219(1–2):273–280
Fong R, von Sacken U, Dahn JR (1990) Studies of lithium intercalation into carbons using nonaqueous electrochemical cells. J Electrochem Soc 137(7):2009–2013
Besenhard JO (1976) The electrochemical preparation and properties of ionic alkali metal-and NR4-graphite intercalation compounds in organic electrolytes. Carbon 14(2):111–115
Gallus DR, Wagner R, Wiemers-Meyer S, Winter M, Cekic-Laskovic I (2015) New insights into the structure-property relationship of high-voltage electrolyte components for lithium-ion batteries using the pKa value. Electrochim Acta 184:410–416
Wagner R, Streipert B, Kraft V, Reyes Jiménez A, Röser S, Kasnatscheew J, Gallus DR, Börner M, Mayer C, Arlinghaus HF (2016) Counterintuitive role of magnesium salts as effective electrolyte additives for high voltage lithium-ion batteries. Adv Mater Interfaces 3(15)
Wagner R, Korth M, Streipert B, Kasnatscheew J, Gallus DR, Brox S, Amereller M, Cekic-Laskovic I, Winter M (2016) Impact of selected LiPF6 hydrolysis products on the high voltage stability of lithium-ion battery cells. ACS Appl Mater Interfaces 8(45):30871–30878
Yazami R, Touzain P (1983) A reversible graphite-lithium negative electrode for electrochemical generators. J Power Sources 9(3):365–371
Basu S (1981) Rechargeable battery. Bell Telephone Laboratories, US Patent 4:304,825
Murmann P, Streipert B, Kloepsch R, Ignatiev N, Sartori P, Winter M, Cekic-Laskovic I (2015) Lithium-cyclo-difluoromethane-1, 1-bis (sulfonyl) imide as a stabilizing electrolyte additive for improved high voltage applications in lithium-ion batteries. Phys Chem Chem Phys 17(14):9352–9358
Ozawa K (1994) Lithium-ion rechargeable batteries with LiCoO2 and carbon electrodes—the LiCoO2/ C system. Solid State Ionics 69(3–4):212–221
Megahed S, Scrosati B (1994) Lithium-ion rechargeable batteries. J Power Sources 51(1–2):79–104
Bieker P, Winter M (2016) Lithium-Ionen-Technologie und was danach kommen könnte. Chem Unserer Zeit 50(3):172–186
Krämer E, Schedlbauer T, Hoffmann B, Terborg L, Nowak S, Gores HJ, Passerini S, Winter M (2013) Mechanism of anodic dissolution of the aluminum current collector in 1 M LiTFSI EC: DEC 3: 7 in rechargeable lithium batteries. J Electrochem Soc 160(2):A356–A360
Krämer E, Passerini S, Winter M (2012) Dependency of aluminum collector corrosion in lithium ion batteries on the electrolyte solvent. ECS Electrochem Lett 1(5):C9–C11
Heckmann A, Krott M, Streipert B, Uhlenbruck S, Winter M, Placke T (2017) Suppression of aluminum current collector dissolution by protective ceramic coatings for better high-voltage battery performance. ChemPhysChem 18(1):156–163
Böttcher T, Duda B, Kalinovich N, Kazakova O, Ponomarenko M, Vlasov K, Winter M, Röschenthaler G-V (2014) Syntheses of novel delocalized cations and fluorinated anions, new fluorinated solvents and additives for lithium ion batteries. Prog Solid State Chem 42(4):202–217
Schmitz RW, Murmann P, Schmitz R, Müller R, Krämer L, Kasnatscheew J, Isken P, Niehoff P, Nowak S, Röschenthaler G-V (2014) Investigations on novel electrolytes, solvents and SEI additives for use in lithium-ion batteries: systematic electrochemical characterization and detailed analysis by spectroscopic methods. Prog Solid State Chem 42(4):65–84
Amereller M, Schedlbauer T, Moosbauer D, Schreiner C, Stock C, Wudy F, Zugmann S, Hammer H, Maurer A, Gschwind R (2014) Electrolytes for lithium and lithium ion batteries: from synthesis of novel lithium borates and ionic liquids to development of novel measurement methods. Prog Solid State Chem 42(4):39–56
Nishi Y (2001) The development of lithium ion secondary batteries. Chem Rec 1(5):406–413
Broussely M, Archdale G (2004) Li-ion batteries and portable power source prospects for the next 5–10 years. J Power Sources 136(2):386–394
Pillot C (2017) The rechargeable battery market and main trends 2016–2025. Talk at Advanced Automotive Battery Conference (AABC) Europe, Mainz
Whittingham MS (2014) Ultimate limits to intercalation reactions for lithium batteries. Chem Rev 114(23):11414–11443
Shao YY, Ding F, Xiao J, Zhang J, Xu W, Park S, Zhang JG, Wang Y, Liu J (2013) Making Li-air batteries rechargeable: material challenges. Adv Funct Mater 23(8):987–1004
Zhang SS (2013) Liquid electrolyte lithium/sulfur battery: fundamental chemistry, problems, and solutions. J Power Sources 231:153–162
Chen L, Shaw LL (2014) Recent advances in lithium-sulfur batteries. J Power Sources 267:770–783
Grande L, Paillard E, Hassoun J, Park J-B, Lee Y-J, Sun Y-K, Passerini S, Scrosati B (2014) The lithium/air battery: still an emerging system or a practical reality? Adv Mater 27(5):784-800
Ogasawara T, Débart A, Holzapfel M, Novák P, Bruce PG (2006) Rechargeable Li2O2 electrode for lithium batteries. J Am Chem Soc 128(4):1390–1393
Hagen M, Hanselmann D, Ahlbrecht K, Maça R, Gerber D, Tübke J (2015) Lithium–sulfur cells: the gap between the state-of-the-art and the requirements for high energy battery cells. Adv Energy Mater 5(16):1401986
Blurton KF, Sammells AF (1979) Metal/air batteries: their status and potential—a review. J Power Sources 4(4):263–279
Abraham KM, Jiang Z (1996) Solid polymer electrolyte-based oxygen batteries. US Patent 5:510,209
Abraham KM, Jiang Z (1996) A polymer electrolyte-based rechargeable lithium/oxygen battery. J Electrochem Soc 143(1):1–5
Choi JW, Aurbach D (2016) Promise and reality of post-lithium-ion batteries with high energy densities. Nature Reviews Materials 1:16013
Danuta H, Juliusz U (1962) Electric dry cells and storage batteries. US Patent 3:043,896
Rao MLB (1966) Organic electrolyte cells. US Patent 3413154 A
Rauh RD, Abraham KM, Pearson GF, Surprenant JK, Brummer SB (1979) A lithium/dissolved sulfur battery with an organic electrolyte. J Electrochem Soc 126(4):523–527
Ji X, Lee KT, Nazar LF (2009) A highly ordered nanostructured carbon-sulphur cathode for lithium-sulphur batteries. Nat Mater 8(6):500–506
Aurbach D, Pollak E, Elazari R, Salitra G, Kelley CS, Affinito J (2009) On the surface chemical aspects of very high energy density, rechargeable Li–sulfur batteries. J Electrochem Soc 156(8):A694–A702
Yin Y-X, Xin S, Guo Y-G, Wan L-J (2013) Lithium–sulfur batteries: electrochemistry, materials, and prospects. Angew Chem Int Ed 52(50):13186–13200
SionPower http://www.sionpower.com (Accessed January 20, 2017)
Yabuuchi N, Kubota K, Dahbi M, Komaba S (2014) Research development on sodium-ion batteries. Chem Rev 114(23):11636–11682
Klein F, Jache B, Bhide A, Adelhelm P (2013) Conversion reactions for sodium-ion batteries. Phys Chem Chem Phys 15(38):15876–15887
Ellis BL, Nazar LF (2012) Sodium and sodium-ion energy storage batteries. Curr Opin Solid State Mat Sci 16(4):168–177
Bachman JC, Muy S, Grimaud A, Chang H-H, Pour N, Lux SF, Paschos O, Maglia F, Lupart S, Lamp P, Giordano L, Shao-Horn Y (2016) Inorganic solid-state electrolytes for lithium batteries: mechanisms and properties governing ion conduction. Chem Rev 116(1):140–162
Hu Y-S (2016) Batteries: getting solid. Nature Energy 1:16042
Weber AZ, Mench MM, Meyers JP, Ross PN, Gostick JT, Liu QH (2011) Redox flow batteries: a review. J Appl Electrochem 41(10):1137–1164
Aurbach D, Weissman I, Gofer Y, Levi E (2003) Nonaqueous magnesium electrochemistry and its application in secondary batteries. Chem Rec 3(1):61–73
Saha P, Datta MK, Velikokhatnyi OI, Manivannan A, Alman D, Kumta PN (2014) Rechargeable magnesium battery: current status and key challenges for the future. Prog Mater Sci 66(0):1–86
Jian Z, Luo W, Ji X (2015) Carbon electrodes for K-ion batteries. J Am Chem Soc 137:11566–11569
Vaalma C, Giffin GA, Buchholz D, Passerini S (2016) Non-aqueous K-ion battery based on layered K0.3MnO2 and hard carbon/carbon black. J Electrochem Soc 163(7):A1295–A1299
Ponrouch A, Frontera C, Barde F, Palacin MR (2016) Towards a calcium-based rechargeable battery. Nat Mater 15(2):169
Reinsberg P, Bondue CJ, Baltruschat H (2016) Calcium-oxygen batteries as a promising alternative to sodium-oxygen batteries. J Phys Chem C 120(39):22179–22185
Wachtler M, Wagner MR, Schmied M, Winter M, Besenhard JO (2001) The effect of the binder morphology on the cycling stability of Li–alloy composite electrodes. J Electroanal Chem 510(1):12–19
Lux S, Schappacher F, Balducci A, Passerini S, Winter M (2010) Low cost, environmentally benign binders for lithium-ion batteries. J Electrochem Soc 157(3):A320–A325
Qi X, Blizanac B, DuPasquier A, Oljaca M, Li J, Winter M (2013) Understanding the influence of conductive carbon additives surface area on the rate performance of LiFePO4 cathodes for lithium ion batteries. Carbon 64:334–340
Qi X, Blizanac B, DuPasquier A, Meister P, Placke T, Oljaca M, Li J, Winter M (2014) Investigation of PF6 − and TFSI− anion intercalation into graphitized carbon blacks and its influence on high voltage lithium ion batteries. Phys Chem Chem Phys 16(46):25306–25313
Qi X, Blizanac B, DuPasquier A, Lal A, Niehoff P, Placke T, Oljaca M, Li J, Winter M (2015) Influence of thermal treated carbon black conductive additive on the performance of high voltage spinel Cr-doped LiNi0.5Mn1.5O4 composite cathode electrode. J Electrochem Soc 162(3):A339–A343
Bockholt H, Haselrieder W, Kwade A (2013) Intensive dry and wet mixing influencing the structural and electrochemical properties of secondary lithium-ion battery cathodes. ECS Trans 50(26):25–35
Bockholt H, Haselrieder W, Kwade A (2016) Intensive powder mixing for dry dispersing of carbon black and its relevance for lithium-ion battery cathodes. Powder Technol 297:266–274
Bauer W, Nötzel D, Wenzel V, Nirschl H (2015) Influence of dry mixing and distribution of conductive additives in cathodes for lithium ion batteries. J Power Sources 288:359–367
Mazouzi D, Karkar Z, Hernandez CR, Manero PJ, Guyomard D, Roue L, Lestriez B (2015) Critical roles of binders and formulation at multiscales of silicon-based composite electrodes. J Power Sources 280:533–549
Porcher W, Lestriez B, Jouanneau S, Guyomard D (2010) Optimizing the surfactant for the aqueous processing of LiFePO4 composite electrodes. J Power Sources 195(9):2835–2843
Du Z, Wood III DL, Daniel C, Kalnaus S, Li J (2017) Understanding limiting factors in thick electrode performance as applied to high energy density Li-ion batteries. J Appl Electrochem 47(3):405–415
Bitsch B, Gallasch T, Schroeder M, Börner M, Winter M, Willenbacher N (2016) Capillary suspensions as beneficial formulation concept for high energy density Li-ion battery electrodes. J Power Sources 328:114–123
Novák P, Scheifele W, Winter M, Haas O (1997) Graphite electrodes with tailored porosity for rechargeable ion-transfer batteries. J Power Sources 68(2):267–270
Haselrieder W, Ivanov S, Christen DK, Bockholt H, Kwade A (2013) Impact of the calendering process on the interfacial structure and the related electrochemical performance of secondary lithium-ion batteries. ECS Trans 50(26):59–70
Antartis D, Dillon S, Chasiotis I (2015) Effect of porosity on electrochemical and mechanical properties of composite Li-ion anodes. J Compos Mater 49(15):1849–1862
Zhang W-J (2011) Lithium insertion/extraction mechanism in alloy anodes for lithium-ion batteries. J Power Sources 196(3):877–885
Zhao H, Yuan W, Liu G (2015) Hierarchical electrode design of high-capacity alloy nanomaterials for lithium-ion batteries. Nano Today 10(2):193–212
Hochgatterer N, Schweiger M, Koller S, Raimann P, Wöhrle T, Wurm C, Winter M (2008) Silicon/graphite composite electrodes for high-capacity anodes: influence of binder chemistry on cycling stability. Electrochem Solid-State Lett 11(5):A76–A80
Vogl U, Das P, Weber A, Winter M, Kostecki R, Lux S (2014) Mechanism of interactions between CMC binder and Si single crystal facets. Langmuir 30(34):10299–10307
Nelson P, Gallagher K, Bloom I BatPaC (battery performance and cost) software, Argonne National Lab, http://www.cse.anl.gov/BatPaC/ (Accessed on January 10, 2017)
Warner J (2015) The handbook of lithium-ion battery pack design—chemistry, components, types and terminology. Elsevier Science, Burlington
3M http://multimedia.3m.com/mws/media/756169O/3mtm-battery-materials.pdf (Accessed March 20, 2017)
Korthauer R (2013) Handbuch Lithium-Ionen-Batterien. Springer Vieweg, Wiesbaden
Kasavajjula U, Wang C, Appleby AJ (2007) Nano- and bulk-silicon-based insertion anodes for lithium-ion secondary cells. J Power Sources 163(2):1003–1039
Obrovac MN, Chevrier VL (2014) Alloy negative electrodes for Li-ion batteries. Chem Rev 114(23):11444–11502
Zhang W-J (2011) A review of the electrochemical performance of alloy anodes for lithium-ion batteries. J Power Sources 196(1):13–24
Qiu B, Zhang M, Xia Y, Liu Z, Meng YS (2017) Understanding and controlling anionic electrochemical activity in high-capacity oxides for next generation Li-ion batteries. Chem Mater 29(3):908–915
Noh H-J, Youn S, Yoon CS, Sun Y-K (2013) Comparison of the structural and electrochemical properties of layered Li[NixCoyMnz]O2 (x = 1/3, 0.5, 0.6, 0.7, 0.8 and 0.85) cathode material for lithium-ion batteries. J Power Sources 233:121–130
Li J, Kloepsch R, Stan MC, Nowak S, Kunze M, Winter M, Passerini S (2011) Synthesis and electrochemical performance of the high voltage cathode material Li[Li0.2Mn0.56Ni0.16Co0.08]O2 with improved rate capability. J Power Sources 196(10):4821–4825
Xia Q, Zhao X, Xu M, Ding Z, Liu J, Chen L, Ivey DG, Wei W (2015) A Li-rich Layered@ Spinel@ Carbon heterostructured cathode material for high capacity and high rate lithium-ion batteries fabricated via an in situ synchronous carbonization-reduction method. J Mater Chem A 3(7):3995–4003
Liu H, Wang J, Zhang X, Zhou D, Qi X, Qiu B, Fang J, Kloepsch R, Schumacher G, Liu Z, Li J (2016) Morphological evolution of high-voltage spinel LiNi0.5Mn1.5O4 cathode materials for lithium-ion batteries: the critical effects of surface orientations and particle size. ACS Appl Mater Interfaces 8(7):4661–4675
Liu N, Lu Z, Zhao J, McDowell MT, Lee H-W, Zhao W, Cui Y (2014) A pomegranate-inspired nanoscale design for large-volume-change lithium battery anodes. Nat Nano 9(3):187–192
Winter M, Besenhard JO, Albering JH, Yang J, Wachtler M (1998) Lithium storage alloys as anode materials for lithium ion batteries. Prog Batt Batt Mater 17:208
Besenhard J, Yang J, Winter M (1997) Will advanced lithium-alloy anodes have a chance in lithium-ion batteries? J Power Sources 68(1):87–90
Qian J, Adams BD, Zheng J, Xu W, Henderson WA, Wang J, Bowden ME, Xu S, Hu J, Zhang J-G (2016) Anode-free rechargeable lithium metal batteries. Adv Funct Mater 26(39):7094–7102
Brückner J, Thieme S, Grossmann HT, Dörfler S, Althues H, Kaskel S (2014) Lithium–sulfur batteries: influence of C-rate, amount of electrolyte and sulfur loading on cycle performance. J Power Sources 268:82–87
Greszler T, Gu W, Goebel S, Masten D, Lakshmanan B (2012) Li-air and Li-sulfur in an automotive system context. Talk at Beyond Lithium Ion 5, Berkeley, CA
Armand M (1994) The history of polymer electrolytes. Solid State Ionics 69(3):309–319
Greatbatch W, Holmes CF (1992) The lithium/iodine battery: a historical perspective. Pacing Clin Electrophysiol 15(11):2034–2036
Vetter J, Novak P, Wagner MR, Veit C, Möller KC, Besenhard JO, Winter M, Wohlfahrt-Mehrens M, Vogler C, Hammouche A (2005) Ageing mechanisms in lithium-ion batteries. J Power Sources 147(1–2):269–281
Seino Y, Ota T, Takada K, Hayashi A, Tatsumisago M (2014) A sulphide lithium super ion conductor is superior to liquid ion conductors for use in rechargeable batteries. Energy Environ Sci 7(2):627–631
Manthiram A, Yu X, Wang S (2017) Lithium battery chemistries enabled by solid-state electrolytes. Nat Rev Mater 2:16103
Pieczonka NPW, Liu Z, Lu P, Olson KL, Moote J, Powell BR, Kim J-H (2013) Understanding transition-metal dissolution behavior in LiNi0.5Mn1.5O4 high-voltage spinel for lithium ion batteries. J Phys Chem C 117(31):15947–15957
Gallus DR, Schmitz R, Wagner R, Hoffmann B, Nowak S, Cekic-Laskovic I, Schmitz RW, Winter M (2014) The influence of different conducting salts on the metal dissolution and capacity fading of NCM cathode material. Electrochim Acta 134:393–398
Börner M, Klamor S, Hoffmann B, Schroeder M, Nowak S, Würsig A, Winter M, Schappacher F (2016) Investigations on the C-rate and temperature dependence of manganese dissolution/deposition in LiMn2O4/Li4Ti5O12 lithium ion batteries. J Electrochem Soc 163(6):A831–A837
Evertz M, Horsthemke F, Kasnatscheew J, Börner M, Winter M, Nowak S (2016) Unraveling transition metal dissolution of Li1.04Ni1/3Co1/3Mn1/3O2 (NCM 111) in lithium ion full cells by using the total reflection X-ray fluorescence technique. J Power Sources 329:364–371
Jia H, Kloepsch R, He X, Evertz M, Nowak S, Li J, Winter M, Placke T (2016) Nanostructured ZnFe2O4 as anode material for lithium ion batteries: ionic liquid-assisted synthesis and performance evaluation with special emphasis on comparative metal dissolution. Acta Chim Slov 63(3):470–483
Xu W, Wang J, Ding F, Chen X, Nasybulin E, Zhang Y, Zhang J-G (2014) Lithium metal anodes for rechargeable batteries. Energy Environ Sci 7(2):513–537
Kato Y, Kawamoto K, Kanno R, Hirayama M (2012) Discharge performance of all-solid-state battery using a lithium superionic conductor Li10GeP2S12. Electrochemistry 80(10):749–751
Gambe Y, Sun Y, Honma I (2015) Development of bipolar all-solid-state lithium battery based on quasi-solid-state electrolyte containing tetraglyme-LiTFSA equimolar complex. Sci Rep 5:8869–8872
Kloepsch R, Placke T, Winter M (2017) Festelektrolytbatterien: Sinn, Unsinn, Realitätssinn. Proceedings, Batterieforum Deutschland, January 25–27, Berlin, Germany
Armand M (1983) Polymer solid electrolytes—an overview. Solid State Ionics 9:745–754
Armand MB (1986) Polymer electrolytes. Annu Rev Mater Sci 16(1):245–261
Baril D, Michot C, Armand M (1997) Electrochemistry of liquids vs. solids: polymer electrolytes. Solid State Ionics 94(1):35–47
Murata K, Izuchi S, Yoshihisa Y (2000) An overview of the research and development of solid polymer electrolyte batteries. Electrochim Acta 45(8–9):1501–1508
Rupp B, Schmuck M, Balducci A, Winter M, Kern W (2008) Polymer electrolyte for lithium batteries based on photochemically crosslinked poly (ethylene oxide) and ionic liquid. Eur Polym J 44(9):2986–2990
Isken P, Winter M, Passerini S, Lex-Balducci A (2013) Methacrylate based gel polymer electrolyte for lithium-ion batteries. J Power Sources 225:157–162
Schroeder M, Isken P, Winter M, Passerini S, Lex-Balducci A, Balducci A (2013) An investigation on the use of a methacrylate-based gel polymer electrolyte in high power devices. J Electrochem Soc 160(10):A1753–A1758
Jankowsky S, Hiller MM, Fromm O, Winter M, Wiemhoefer H-D (2015) Enhanced lithium-ion transport in polyphosphazene based gel polymer electrolytes. Electrochim Acta 155:364–371
Bruce PG, West AR (1983) The A-C conductivity of polycrystalline LISICON, Li2+2x Zn1-x GeO4, and a model for intergranular constriction resistances. J Electrochem Soc 130(3):662–669
Aono H, Sugimoto E, Sadaoka Y, Imanaka N, Adachi G (1990) Ionic-conductivity of solid electrolytes based on lithium titanium phosphate. J Electrochem Soc 137(4):1023–1027
Inaguma Y, Chen LQ, Itoh M, Nakamura T, Uchida T, Ikuta H, Wakihara M (1993) High ionic-conductivity in lithium lanthanum titanate. Solid State Commun 86(10):689–693
Murugan R, Thangadurai V, Weppner W (2007) Fast lithium ion conduction in garnet-type Li7La3Zr2O12. Angew Chem, Int Ed 46(41):7778–7781
Yu XH, Bates JB, Jellison GE, Hart FX (1997) A stable thin-film lithium electrolyte: lithium phosphorus oxynitride. J Electrochem Soc 144(2):524–532
Wang Y, Richards WD, Ong SP, Miara LJ, Kim JC, Mo YF, Ceder G (2015) Design principles for solid-state lithium superionic conductors. Nat Mater 14(10):1026
Sakuda A, Hayashi A, Tatsumisago M (2013) Sulfide solid electrolyte with favorable mechanical property for all-solid-state lithium battery. Sci Rep 3:2261
Muramatsu H, Hayashi A, Ohtomo T, Hama S, Tatsumisago M (2011) Structural change of Li2S–P2S5 sulfide solid electrolytes in the atmosphere. Solid State Ionics 182(1):116–119
Kamaya N, Homma K, Yamakawa Y, Hirayama M, Kanno R, Yonemura M, Kamiyama T, Kato Y, Hama S, Kawamoto K, Mitsui A (2011) A lithium superionic conductor. Nat Mater 10(9):682–686
Wenzel S, Randau S, Leichtweiss T, Weber DA, Sann J, Zeier WG, Janek J (2016) Direct observation of the interfacial instability of the fast ionic conductor Li10GeP2S12 at the lithium metal anode. Chem Mater 28(7):2400–2407
Wenzel S, Weber DA, Leichtweiss T, Busche MR, Sann J, Janek J (2016) Interphase formation and degradation of charge transfer kinetics between a lithium metal anode and highly crystalline Li7P3S11 solid electrolyte. Solid State Ionics 286:24–33
Zhu YZ, He XF, Mo YF (2016) First principles study on electrochemical and chemical stability of solid electrolyte-electrode interfaces in all-solid-state Li-ion batteries. J Mater Chem A 4(9):3253–3266
Metalary http://metalary.com/lithium-price/. Accessed 8 March 2017
Cekic-Laskovic I, Wagner R, Wiemers-Meyer S, Nowak S, Winter M (2016) Liquid electrolytes—just a commodity and a phase-out model? Proceedings, Graz Battery Days, September 26–28, Graz, Austria
Bieker G, Winter M, Bieker P (2015) Electrochemical in situ investigations of SEI and dendrite formation on the lithium metal anode. Phys Chem Chem Phys 17(14):8670–8679
Ryou MH, Lee YM, Lee Y, Winter M, Bieker P (2015) Surface treatment: mechanical surface modification of lithium metal: towards improved Li metal anode performance by directed Li plating. Adv Funct Mater 25(6):825–825
Martha SK, Nanda J, Kim Y, Unocic RR, Pannala S, Dudney NJ (2013) Solid electrolyte coated high voltage layered-layered lithium-rich composite cathode: Li1.2Mn0.525Ni0.175Co0.1O2. J Mater Chem A 1(18):5587–5595
Li XF, Liu J, Banis MN, Lushington A, Li RY, Cai M, Sun XL (2014) Atomic layer deposition of solid-state electrolyte coated cathode materials with superior high-voltage cycling behavior for lithium ion battery application. Energy Environ Sci 7(2):768–778
Woodford WH, Carter WC, Chiang Y-M (2012) Design criteria for electrochemical shock resistant battery electrodes. Energy Environ Sci 5(7):8014–8024
Acknowledgements
The authors wish to thank the German Ministry of Education and Research (BMBF) for funding this work in the project “BenchBatt” (03XP0047A). The authors also want to thank Andre Bar for the preparation of various graphics for this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Placke, T., Kloepsch, R., Dühnen, S. et al. Lithium ion, lithium metal, and alternative rechargeable battery technologies: the odyssey for high energy density. J Solid State Electrochem 21, 1939–1964 (2017). https://doi.org/10.1007/s10008-017-3610-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-017-3610-7