
Abstract
We demonstrate the Laplace transform (LT) impedance technique for measuring electrochemical lithiation and delithiation in the two-phase system of spinel Li1 + x Mn2O4 (0 ≤ x ≤ 1). Square constant charge or discharge current pulses with various current densities are applied to the equilibrated Li1.25Mn2O4 system, and the response overpotential is recorded with various sampling rates. The LT of the current and overpotential as a function of time gives frequency-dependent impedance spectra. The results show asymmetric impedance between charge and discharge. In particular, inductive loop resistance, which may stem from the nucleation and growth mechanisms, is visible for mainly the anodic (charging) process. The LT impedance is fitted by a complex non-linear least squares technique. The resulting separated resistances decrease with current density in the lower frequency region, indicating non-ohmic Butler–Volmer-type behavior.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Research and development of spinel LiMn2O4 for positive electrode in rechargeable Li-ion batteries are vital; LiMn2O4 is cheap, and its mineral resources are abundant compared with widely used LiCoO2 and related ceramics [1–4]. A major obstacle to obtaining high energy densities in Li1 + x Mn2O4 is poor cycle and rate performance in the compositional range of x = 0 to 1, and thus only half of its theoretical capacity is available (148 mAh/g for Li1 + x Mn2O4 (0 ≤ (1 + x) ≤ 1)). In general, this poor performance arises from the phase transition from the cubic to the tetrahedral Li2Mn2O4 phase via a two-phase reaction and the Jahn–Teller distortion effect of Mn3+ ions. Okubo et al. improved the rate performance by controlling the particle size, and the two-phase interface of the LiMn2O4 and Li2Mn2O4 phases in the particles plays a crucial role in the kinetics of processes, such as nucleation and growth [5].
Therefore, quantitative studies of reaction kinetics are essential to understanding and improving the reaction rates, particularly for the two-phase system. The AC impedance technique is widely used to evaluate the kinetics of various elemental reactions and processes, such as ohmic resistances (electronic conduction in electrode composites and ionic conductivity in electrolyte solutions), charge transfer reactions at the interface between the electrode and the electrolyte and diffusion of lithium in solid electrode particles or in liquid electrolyte in the micropores or mesopores of the electrode composite [6]. In particular, the mechanism of interfacial charge transfer reactions is still uncertain. Recently, in a study using AC impedance measurements and ab initio density functional theory calculations, we suggested that in Li1 + x Mn2O4 (0 ≤ (1 + x) ≤ 1), two processes are involved in the interfacial reactions, namely desolvation and lattice incorporation, owing to the chemical potential gradient at the electrode surface [7, 8]. However, there are technical limitations to typical AC impedance measurements. The method cannot distinguish charge and discharge reactions because of AC bias, and it is hard to evaluate the electrochemical polarization dependence as a function of current density. The electrochemical polarizations of the interfacial charge transfer reaction and diffusion of Li are non-ohmic. Therefore, direct polarization measurements under high-rate conditions are necessary, particularly for electric vehicle batteries, which require high-rate performance. In addition, the two-phase reaction may include nucleation and growth (domain boundary moving) processes that do not occur in solid solution systems. Nucleation and growth mechanisms may be difficult to distinguish and evaluate by AC impedance measurements, because small perturbations of AC voltage amplitude are not sufficient to exceed oversaturation or to form nucleation embryos. However, DC-based electrochemical techniques are unconstrained by the drawbacks of AC impedance measurements. Ohkubo et al. used the potential intermittent titration technique (PITT) for the Li x Mn2O4 (1 ≤ x ≤ 2) system and evaluated the activation energy of the moving two-phase boundaries in the particle based on the core-shell phase separation in the single particle and considering nucleation and growth mechanisms developed with the Johnson–Mehl–Avrami model [5, 9–11]. In this model, the effective overall reaction rate containing the speed of the domain boundary movement was quantitatively evaluated, which may appear in the low-frequency region. However, Bai et al. suggested a mathematical model, in which phase-transforming nanoparticles in porous electrodes consist of lithiated and delithiated particles as the stable phase and active particles as the intermediate transition state for the LiFePO4/FePO4 system [12]. Further investigation requires alternative electrochemical methods because PITT may not be able to distinguish elemental reactions and processes in the high- and middle-frequency regions arising from processes such as nucleation and charge transfer reactions.
Recently, Huang et al. proposed an improved AC impedance measurement technique under charge (discharge) current flow and used the technique to measure the electrochemical reaction of Li1 + x Mn2O4 spinel compounds [13, 14]. However, the method is limited to higher frequency regions corresponding to the range of the current pulse duration.
Takano et al. proposed impedance spectroscopy using the Laplace transform (LT) for the time-dependent response current (voltage) profile to DC voltage (current) perturbation [15, 16]. This technique may be a promising alternative to small-amplitude AC sine-wave voltage perturbation. Conventional fitting procedures based on equivalent circuits can also be used for the LT impedance spectra, and thus the method can analyze the electrochemical polarization behavior of the charge or discharge reaction separately as a function of current density.
In this study, the electrochemical polarization of the two-phase reaction in Li1 + x Mn2O4 (0 ≤ x ≤ 1) is evaluated from the LT impedance spectra as a function of current density (C-rate) and its dependence on the charge or discharge reaction. We mainly focus on using the LT impedance method to observe the electrochemical behavior of the two-phase reaction phenomenologically; thus, clarifying the elemental reaction mechanism is beyond the scope of this study.
Experimental
The LiMn2O4 samples were prepared by a conventional solid-state reaction. Mn2O3 was prepared by preheating MnCO3 at 600 °C for 24 h. A stoichiometric mixture of Li2CO3 and Mn2O3 was heated at 820 °C for 3 days in air and then cooled at a rate of 0.5 °C/min. The phase and the lattice parameters were determined by powder X-ray diffraction (XRD; MiniFlex 600, Rigaku Co., Ltd.) with Cu Kα radiation. Particle morphology and size were evaluated by scanning electron microscopy (SEM) and nitrogen gas adsorption/desorption isotherm measurements with Brunauer–Emmett–Teller (BET) theory.
The slow speed galvanostatic electrochemical Li+ insertion and open circuit voltage (OCV) were measured with a three-electrode cell. The working electrode was a mixture of 70 wt% sample powder, 25 wt% acetylene black (AB), and 5 wt% poly (tetrafluoroethylene) binder. Li foil (Aldrich) was used as the counter and reference electrode, and 1 M LiClO4 in ethylene carbonate (EC) and diethylene carbonate (DEC) was used as the electrolyte (EC/DEC = 1:1, Tomiyama Pure Chemical, Ltd.). Electrode sheets used for DC galvanostatic pulse cycle measurements consisted of an active material (LiMn2O4), AB as an electronic conductive agent, and poly (vinylidene fluoride) as a binder at a weight ratio of 80:10:10. The mixture was ball milled at 2000 rpm for 15 min with agate milling balls. A lithium metal counter electrode and 1 M LiClO4 in EC/DEC = 1/1 (v/v) electrolyte solution were used. We assembled airtight three-electrode beaker cells and two-electrode coin cells in an Ar-filled glove box.
The electrochemical tests were performed at 20 °C with a multichannel potentio-galvanostat equipped with impedance modules (VMP3, Bio-Logic). OCV measurements were carried out with a galvanostatic electrochemical Li insertion at a rate of 0.05 C followed by a sufficient relaxation time of 12 h. DC galvanostatic pulse cycle measurements for LT impedance measurements were performed by using the method in our previous study [17]. The composition was x = 1.25 in Li x Mn2O4, which was obtained by galvanostatic Li insertion and an 8-h relaxation under open circuit conditions. Galvanostatic cathodic and anodic (discharge and charge) square-wave pulses were alternately applied at various current densities from 0.3 to 2.0 C. The duration of the square-wave pulse was 20 s (<2 C) or 10 s (for 2.0 C), with intermittent relaxation for 8 h (galvanostatic cycles). The response voltage profile against time, v(t), was recorded by a four-channel logger with various sampling rates in order to cover various frequency range. In detail, the sampling rates are 1000, 100, 10, and 1 points per second with a sampling duration of 50, 180, 1200, and 28,800 s, respectively. The electrochemical polarization, η(t), was calculated by subtracting OCV from the response voltage, v(t).
The LTs, I(s) and V(s), of the inputted current and response polarization, i(t) and η(t), are calculated as
where s is the Laplace parameter expressed as
where j is an imaginary unit, (−1)1/2, a is a positive real constant, a = 2πj/T, ω is the angular frequency, and T is the analyzing period according to the literature [15, 16]. As a result, the Laplace impedance function, Z L(s), is defined as
The frequency-dependent complex impedance, Z(ω), is approximated by a first-order Maclaurin expansion as
The details are described in the literature [15, 16]. Note that frequency range of LT impedance method, in principle, depends on the sampling rate and total sampling period for higher and lower frequency ends, respectively. Technically, increasing of sampling rate leads to scattering of each voltage response measurement. Since LT impedance technique is sensitive to noise signal, the high frequency end was set as 100 Hz in the present experimental condition, using Li/LiMn2O4 two-electrode coin-type cell. In addition, we set the minimum current density as 0.1 C in order to obtain sufficiently qualified spectra.
Results and discussion
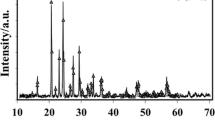
Figure 1a, b shows the powder XRD patterns and SEM images of synthesized LiMn2O4, respectively. The images confirm the formation of the spinel structure (space group: Fd-3m), where Li and Mn ions occupy tetrahedral 8a and octahedral 16d sites, respectively. The grain size of ~0.5 μm obtained from the SEM image is also supported by the BET measurements (0.475 ± 0.02-μm diameter, Fig. S-1 in the supplementary material).

XRD pattern (a) and SEM images (b) of LiMn2O4
Figure 2 shows the voltage profile of Li1 + x Mn2O4 (0 ≤ x ≤ 0.8) as a function of composition x obtained by the galvanostatic intermittent titration technique (GITT) using a three-electrode cell. OCVs obtained after 12-h relaxation are constant regardless of composition x, indicating two coexisting reactions. The electrochemical polarization increases rapidly at x > 0.7, despite the constant OCV. This agrees qualitatively with the previous report by Okubo et al. [5], owing to slow diffusion in the core-shell configuration of Li-rich and Li-poor phases in the particle. Therefore, we set the initial composition as x = 0.25 for the galvanostatic pulse cycles to avoid the slow diffusion effect.

Variation of the voltage with Li+ insertion as a function of composition x in Li1 + x Mn2O4 by GITT with a three-electrode cell. Lithium metal is used for the reference electrode. Filled circles represent the OCV after an 8-h relaxation
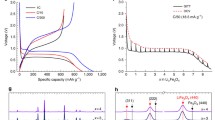
The measured DC electrochemical polarization profiles as a function of time are presented in Fig. 3a. A coin-type two-electrode cell is used for the measurement. The duration of the corresponding galvanostatic square wave is 10 (for 2 C) or 20 s (<2 C). The polarization profiles at various C-rate pulses are obtained by cycling charge/discharge pulse sequence. Conventional AC impedance measurements (10 mV for AC amplitude and 10 mHz–105 Hz for the frequency range after relaxation) were performed before and after charge/discharge pulse cycles (Fig. 3b), and no marked difference is observed in the spectra. Thus, it is confirmed that were no degradation of electrochemical reaction during the charge/discharge pulse cycles.

a Typical polarization profile for a galvanostatic charge–discharge pulse. The deviation from the OCV value corresponds to polarization for charge (if the polarization is positive) and discharge (negative). b Electrochemical AC impedance spectroscopy before and after galvanostatic charge–discharge pulse cycles in a two-electrode Li/Li1 + x Mn2O4 cell
The lower C-rate pulse shows symmetric electrochemical polarization profiles against the OCV (0.1 C), indicating no marked difference between charge and discharge polarization processes. On the other hand, higher C-rate pulse shows asymmetric electrochemical polarization profiles, i.e., different charge/discharge polarization mechanisms. In detail, at a low rate of 0.1 C, an abrupt increase in the electrochemical polarization at the beginning of the voltage response is followed by a monotonic increase in the polarization as a function of time for both the charge and discharge pulses. This can be explained by the conventional interpretation of electrochemical reactions and processes. The initial abrupt increase of electrochemical polarization in Fig. 3 mainly stems from the ohmic resistance, whereas the gradual increase in the polarization arises from the diffusion process. The ion exchange reaction covers the region between the ohmic resistance and diffusion processes, and thus separating the three types of electrochemical polarization is technically difficult. In addition to these common features between charge and discharge, a momentum increase in the electrochemical polarization immediately after the abrupt increase caused by ohmic resistance is visible as a response to a higher C-rate charge (anodic) current pulse (>1 C). This momentum increase in the voltage profile indicates deviation from the lowest energy path based on bulk thermodynamics. An example is a nucleation-driven kinetics. Figure 4a shows the LT impedance spectra (100 of the obtained polarization profiles at the charging current density of 0.1 C and corresponding AC impedance spectra (10 mV for AC amplitude and 10 mHz–105 Hz for the frequency range after relaxation). The LT impedance spectra is slightly scattered above 100 Hz due to inclusion of noisy signal in response voltage profile at high-sampling rate measurement. The arks at the lower frequency region than 100 Hz are close each other, indicating almost linear (ohmic) response of voltage profile for the current density of 0.1 C. On the other hand, a large difference between LT and AC impedance spectra is indicated at the high current density of 2.0 C as shown in Fig. 4b, c. In detail, the real part of the LT impedance (Z’) at the highest frequency is similar to that of the AC impedance at the corresponding frequency for all the current density conditions. Therefore, there are no major differences between the LT and the AC impedance spectra in the higher frequency region (>100 Hz). However, the LT impedance in the lower frequency region (<100 Hz) is smaller than the AC impedance and decreases with current density. Therefore, the results indicate a non-ohmic effect against current density in the lower frequency region. The semicircle in the lower frequency region (0.2–6 Hz) is observed in the anodic and cathodic LT impedance spectra. These semicircles may arise from the ion exchange process at the electrode surface, or, more specifically, the lattice incorporation mechanism, because the frequency region is the same as that reported in previous studies [7, 8, 18]. In addition, the inductive loop resistance, which is negligible in AC impedance measurements [13], is visible for the LT impedance spectra under the anodic and higher current density conditions. These characteristic inductive loops are ascribed to the momentum increase in the electrochemical polarization profile during charging (Fig. 3) due to decreasing of polarization (response voltage) profile with time, i.e., phase lead component in impedance spectra [19, 20].

LT impedance spectra around x ~0.25 in Li1 + x Mn2O4 at a rate of 2 C for charge (a) and discharge (b) (filled circles and solid red line). For comparison, the AC impedance spectra after the corresponding galvanostatic pulse charge and discharge measurements are plotted as well (open triangles and dashed line)
Based on these observations, we propose a model equivalent circuit (Fig. 5a). The resistances, R 1 and R 2, are the ohmic resistances due to ion or electron conduction, solid electrolyte interphase (SEI) resistance, and desolvation. In this study, separating the R 1 and R 2 resistance is difficult because the highest frequency of the LT impedance is 100 Hz, and thus it is beyond the scope of this study. Therefore, these impedances are fitted as a single variable, R 1 + R 2. The semicircles at 0.2–6 Hz are represented by an RQ parallel circuit consisting of a resistor, R 3, and a constant phase element (CPE), Q 3. The resistance of the inductive loop is described by the RL parallel circuit (L 4 and R 4). The RL parallel circuit is omitted for fitting the polarization profiles, unless the momentum increase is observed at the onset of charge. The inductive loops are hardly visible for the lithiation process. Therefore, we use an alternative equivalent circuit (Fig. 5b), where CPE Q 4 is used instead of inductor L 4 in Fig. 5a. Lastly, the resistance in the lowest frequency region, R 5, may represent Li+ diffusion at the electrode sheet or phase boundary movement for the two-phase reaction [11], and this is mimicked by the RQ parallel circuit (R 5 and Q 5). Based on the equivalent circuits in Fig. 5a, b, the complex non-linear least squares (CNLS) method [21] is used to evaluate the parameters of the circuit elements quantitatively. The resulting chi-squared functions (χ 2) (less than 10−3 for all spectra) are slightly higher than for the conventional AC impedance technique, because the end of the sampling frequency range of each recorder tends to show a high noise level. However, the measured plots and fitted curves show reasonable agreement (Fig. 5c, d).

Equivalent circuits used for curve fitting of the observed impedance spectra. a Circuit used mainly for anodic polarizations under high current density conditions, which shows a momentum increase in the polarization profile. b Circuit used for cathodic polarization. Typical fitting results for the spectra obtained at a rate of 0.8 C (c) under anodic polarization (charging) and at a rate of 0.5 C (d) under cathodic polarization (discharging). Solid symbols represent observed LT impedance spectra, and dashed lines represent fitted curves
Figure 6 plots the fitted polarization, η 1–η 4, as a function of the current corresponding to the current density range of 0.2–1.2 C. The polarization, η 1–η 4, corresponds to the product of the current and resistances, R 1–R 4, respectively. As mentioned before, R 1 and R 2 cannot be separated, because the sampling rate for the response voltage recording did not cover the corresponding frequency range. Therefore, the sum of R 1 + R 2 is used for the plots. The polarization of η 1 + η 2 shows a linear ohmic relationship with the current. This implies that the main part of R 1 + R 2 arose from the electronic or ionic conduction in the electrode sheet or SEI resistance. However, the AC impedance measurement (Fig. 4) shows a single semicircle in the high-frequency region (>100 Hz), which corresponds to the desolvation process [7, 18]. Because desolvation proceeds in the liquid phase regardless of the chemical potential of Li at the solid-state electrodes, we infer that the resistance shows roughly ohmic behavior in the present current density range. However, R 3 and R 4 decrease non-linearly with the current density, indicating deviation from ohmic behavior. According to the literature [14] and Butler–Volmer kinetics, the relationship between current density, i, and polarization, η, can be described by

Decomposed polarizations, η 1–η 4, by LT impedance analyses (Fig. 5) as a function of current for the galvanostatic pulse cycles with polarization under an anodic (a) and cathodic (b) current. Solid lines are fitted lines or curves. η 1 + η 2 is linear, indicating ohmic behavior, whereas the polarizations of η 3 and η 4 are fitted based on Butler–Volmer kinetics
where α is the transference coefficient, i 0 is the exchange current density (surface area dependent), n is the number of electrons for the elemental electrochemical reaction (n = 1 for Li insertion/removal), F is the Faraday constant, k B is the Boltzmann constant, N is the Avogadro number, and T is the temperature. CNLS fitting is used, and a typical example of a fitted curve is compared with the experimental results in Fig. 6, which show reasonable agreement for polarizations η 3 and η 4. In addition, the fitted transfer coefficients, α, for R 3 and R 4 are ~0.3 and ~0.2 for charging, respectively and that of R 3 and R 4 are ~0.4 and 0.6 for discharging, respectively. Since it is empirically known that transfer coefficients are typically converged to α ~0.5 in many measurements, the transfer coefficients for charge reaction indicate strong asymmetry in their reaction mechanism. So far, the electrochemical reaction mechanism for R 3 and R 4 is uncertain yet, which is beyond the scope of this study as mentioned in the introduction section. However, we think that the observation of inductive loop for charging process may be a clue to precise understanding of the reaction mechanism. For example, Oyama et al. [11] and we [17] proposed nucleation and growth processes, whereas Bai et al. [22] explained the phenomena by the simulation assuming the intermediate phase formation in the porous cathode composite morphology, for LiFePO4-positive electrode. Further investigations for various electrodes are planned to clarify the origin of asymmetry.
Summary
Electrochemical measurements using an LT impedance technique at various current densities were used to analyze the reaction dynamics in a Li-ion battery to separate the charge and discharge reactions and to evaluate non-ohmic behavior under high-rate conditions quantitatively. The lithiation and delithiation processes in the two-phase system of spinel Li1 + x Mn2O4 (0 < x < 1) were investigated. We confirmed the asymmetry of the charge and discharge reactions, and the momentum increase in the electrochemical polarization may be caused by the nucleation and growth mechanisms. Good fitting results for the current density dependence of the observed resistance based on Butler–Volmer kinetics indicated a thermal activation process for resistance R 4, which shows an inductive loop. It is still controversial whether R 4, which corresponds to the momentum increase in the electrochemical polarization in Fig. 6, is the nucleation/growth mechanism in intraparticle or interparticle phase separation [11, 23] or the phase transformation kinetics in a porous electrode containing active particles [22]. We plan to investigate the elemental reaction process in the two-phase reaction by measuring the LT impedance dependence on the electrolyte solution, lithium composition, doping effect, and temperature.
References
Thackeray MM (1997) Manganese oxides for lithium batteries. Progress in Solid State Chemistry 25(1–2):1–71
Ohzuku T, Kitagawa M, Hirai T (1990) Electrochemistry of manganese dioxide in lithium nonaqueous cell III. X-ray diffractional study on the reduction of spinel-related manganese dioxide. J Electrochem Soc 137:769
Guyomard D, Tarascon JM (1994) The carbon/Li1+xMn2O4 system. Solid State Ionics 69:222
Masquelier C, Tabuchi M, Ado K, Kanno R, Kobayashi Y, Maki Y, Nakamura O, Goodenough JB (1996) Chemical and magnetic characterization of spinel materials in the LiMn2O4-Li2Mn4O9-Li4Mn5O12 system. J Solid State Chem 123(2):255–266
Okubo M, Mizuno Y, Yamada H, Kim J, Hosono E, Zhou H, Kudo T, Honma I (2010) Fast Li-ion insertion into nanosized LiMn2O4 without domain boundaries. ACS Nano 4(2):741–752
Macdonald JR (1992) Impedance spectroscopy. Ann Biomed Eng 20(3):289–305
Nakayama M, Taki H, Nakamura T, Tokuda S, Jalem R, Kasuga T (2014) Combined computational and experimental study of Li exchange reaction at the surface of spinel LiMn2O4 as a rechargeable Li-ion battery cathode. J Phys Chem C 118(47):27245–27251
Bruce PG, Saidi MY (1992) The mechanism of electrointercalation. J Electroanal Chem 322:93
Allen JL, Jow TR, Wolfenstine J (2007) Kinetic study of the electrochemical FePO4 to LiFePO4 phase transition. Chem Mater 19(8):2108–2111
Allen JL, Jow TR, Wolfenstine J (2008) Analysis of the FePO4 to LiFePO4 phase transition. J Solid State Electrochem 12:1031
Oyama G, Yamada Y, Natsui R-i, Nishimura S-i, Yamada A (2012) Kinetics of nucleation and growth in two-phase electrochemical reaction of LixFePO4. J Phys Chem C 116(13):7306–7311
Bai P, Cogswell DA, Bazant MZ (2011) Suppression of phase separation in LiFePO4 nanoparticles during battery discharge. Nano Lett 11(11):4890–4896
Huang J, Ge H, Li Z, Zhang J (2015) Dynamic electrochemical impedance spectroscopy of a three-electrode lithium-ion battery during pulse charge and discharge. Electrochim Acta 176:311–320
Huang J, Zhang J, Li Z, Song S, Wu N (2014) Exploring differences between charge and discharge of LiMn2O4/Li half-cell with dynamic electrochemical impedance spectroscopy. Electrochim Acta 131:228–235
Takano K, Nozaki K, Saito Y, Kato K, Negishi A (2000) Impedance spectroscopy by voltage-step chronoamperometry using the Laplace transform method in a lithium-ion battery. J Electrochem Soc 147(3):922–929
Takano K, Nozaki K, Saito Y, Negishi A, Kato K, Yamaguchi Y (2000) Simulation study of electrical dynamic characteristics of lithium-ion battery. J Power Sources 90(2):214–223
Nakayama M, Iizuka K, Shiiba H, Baba S, Nogami M (2011) Asymmetry in anodic and cathodic polarization profile for LiFePO4 positive electrode in rechargeable Li ion battery. J Ceram Soc Jpn 119(1393):692–696
Nakayama M, Ikuta H, Uchimoto Y, Wakihara M (2003) Study on the AC impedance spectroscopy for the Li insertion reaction of LixLa1/3NbO3 at the electrode-electrolyte interface. J Phys Chem B 107(38):10603–10607
Yadav DK, Chauhan DS, Ahamad I, Quraishi MA (2013) Electrochemical behavior of steel/acid interface: adsorption and inhibition effect of oligomeric aniline. RSC Adv 3(2):632–646
Keddam M, Kuntz C, Takenouti H, Schustert D, Zuili D (1997) Exfoliation corrosion of aluminium alloys examined by electrode impedance. Electrochim Acta 42(1):87–97
Macdonald JR, Johnson WB (2005) Fundamentals of impedance spectroscopy. In: Impedance spectroscopy. Wiley, Inc., pp 1–26
Bai P, Tian G (2013) Statistical kinetics of phase-transforming nanoparticles in LiFePO4 porous electrodes. Electrochim Acta 89:644–651
Orvananos B, Yu HC, Abdellahi A, Malik R, Grey CP, Ceder G, Thornton K (2015) Kinetics of nanoparticle interactions in battery electrodes. J Electrochem Soc 162(6):A965–A973
Acknowledgments
This work was partially supported by the JST PRESTO program, a Grant-in-Aid for Scientific Research (no. 257959) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, and the “Elements Strategy Initiative to Form Core Research Center” (since 2012) of MEXT.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(DOCX 22 kb.)
Rights and permissions
About this article

Cite this article
Nakayama, M., Nishimura, N., Kondo, Y. et al. Laplace transform impedance analysis in the two-phase coexistence reaction of spinel Li1 + x Mn2O4 positive electrode. J Solid State Electrochem 21, 1137–1143 (2017). https://doi.org/10.1007/s10008-016-3465-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-016-3465-3