
Abstract
Thermodynamic properties of solid phases in the Cu–O–Al2O3 system were measured by means of the EMF method with oxygen concentration galvanic cells based on stabilized zirconia solid electrolytes. The standard Gibbs energies of formation of pure copper oxides and copper aluminates from their component oxides were determined. Copper aluminates were also investigated calorimetrically by the DSC-TGA combined techniques. Based on the calorimetric measurements, the enthalpies and temperatures of spinel decomposition and delafossite melting were determined. The obtained results were discussed and compared with the available literature data.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Accurate thermodynamic data in the Cu–O–Al2O3 system are essential in pyrometallurgical copper-making processes, in both the primary smelting and the secondary processing where aluminum can be present in raw materials with significant concentration [1]. Besides that, a possible future need for Cr-free refractories as an alternative to the widely used magnesia-chrome due to its potential hazardous features can lead to developing new brick types [2]. A petrographic examination of a worn high-Al2O3 brick taken from a copper anode furnace door revealed formation of CuAlO2 compound [3]. Thus, the conditions under which alumina-rich refractory bricks react with copper melts are of great interest [4].
Thermodynamics of the Cu–O–Al2O3 system and its phase relations are also important in the preparation of Cu/Al2O3 composites with different metal-ceramic bonding techniques, such as eutectic bonding, solid-state bonding, and internal oxidation. Particularly, CuAlO2 plays an important role in the bonding processes [5–8].
Solid phases of the Cu–O–Al2O3 system have a wide range of applications. Due to specific electrical, magnetic, structural, optical, and catalytic properties, copper and its oxides have attracted considerable attention. Copper is widely used as a conductor due to its high electrical and thermal conductivity. Copper oxides are applied in high-temperature superconductors and magnetic storage media, in catalysis and battery applications as well as for gas sensing. CuO and Cu2O are p-type semiconductors with a low band gap of 1.2 and 2.2 eV, respectively [9]. CuAlO2 is also a p-type semiconductor with a band gap wider than in Cu2O, and it belongs to transparent conducting oxides (TCOs) valuable for infrared reflective coatings and for optical displays [10]. CuAl2O4 is used as a catalyst for the hydrogen production and for the catalytic conversion of CO, as a stabilizer of Cu-laden sludges, in the hydrogenolysis of glycerol from biomass, and for CO2 capturing in the chemical-looping combustion [11].
The Cu–O system
The ternary Cu–O–Al2O3 system, which is the subject of the present research, is comprised of three binaries, while the complexity of the ternary system is mainly associated with the Cu–O binary. Due to the practical importance of the Cu–O system, several experimental works and computational studies [12–14] have been reported in the literature. The most recent one is a comprehensive critical assessment of the Cu–O system by Shishin and Decterov [15], where calculated phase diagrams of the Cu–O system were presented based on the reassessed data. In the solid state, the system has two oxides, Cu2O and CuO, thermodynamic stabilities of which are obviously functions of temperature and oxygen partial pressure. For instance, in air, CuO decomposes spontaneously into Cu2O with oxygen evolution at 1299 K [16–20].
The Cu–O–Al2O3 system
In addition to the two well-known copper oxides, two thermodynamically stable ternary phases exist in the Cu–O–Al2O3 system, i.e., a delafossite structured cuprous aluminate, CuAlO2, and copper aluminate spinel, CuAl2O4. Their thermodynamic properties as well as phase relationships in the Cu–O–Al2O3 system studied in the previous works are summarized below.
Schmalzried measured the standard Gibbs energy of formation of CuAlO2 from its component oxides in an electrochemical study with a solid electrolyte oxygen concentration cell at 1273 K [21]. Misra and Chaklader [22] investigated the copper oxide-alumina system in air and developed the phase diagram from 873 to 1973 K by studying prefired samples of mixed CuO and Al2O3 with different ratios and applying X-ray diffraction (XRD) and microscopy to the equilibrated quenched samples. Gadalla and White [23] investigated the CuO–Cu2O–Al2O3 system by the thermogravimetric analysis (TGA) in a temperature range from 1273 to 1573 K at various oxygen partial pressures from 0.21 to 1 atm and developed the Al2O3–(CuO + ½Cu2O) projected isobaric sections, as well as the liquidus surface of the CuO–½Cu2O–Al2O3 system. In addition to that, several reaction equilibria occurring in the Cu–CuO–Al2O3 system were calculated based on the T-P O2 relationships determined for the monovariant transitions at the solid state. Particularly, the standard Gibbs energies of formation of the copper aluminates from their respective component oxides were calculated. Navrotsky and Kleppa measured the enthalpy of formation of CuAl2O4 from its component oxides by a solution calorimetry in a molten oxide solvent at 970 K [24]. Zalazinskii et al. determined thermodynamic stability of CuAlO2 from 973 to 1273 K by reduction with hydrogen, where composition of the equilibrium gas phase was analyzed [25]. Slobodyanyuk et al. reported thermodynamic stabilities of the solid phases in the Cu–CuO–Al2O3 system determined by oxygen concentration galvanic cells and a high-temperature XRD analysis within the temperature range from 1073 to 1273 K [26]. Jacob and Alcock measured the standard Gibbs energy of formation of CuAlO2 and CuAl2O4 by means of the electromotive force (EMF) method and refined the previously reported phase equilibria data in the Cu2O–CuO–Al2O3 system [4].
Tsuchida et al. employed a combined technique of TGA and differential thermal analysis (DTA) with a support of high-temperature XRD in their thermoanalytical study to determine the reaction processes occurring in the copper oxide-alumina system in air up to 1573 K. Particular attention was given to the reactivity of different polymorphs of alumina [27]. Trumble analyzed conditions of CuAlO2 formation at Cu–Al2O3 interfaces based on the available thermodynamic data and presented an assessed isothermal section of the Cu–O–Al system at 1273 K [28]. Also, an invariant point was calculated and experimentally verified, where Cu(l), Cu(s), Al2O3(s), CuAlO2(s), and the gas phase coexist at 1348 K and an oxygen partial pressure of 5.5 × 10−7 atm [6, 29]. Guedes et al. employed a combined technique of TGA and differential scanning calorimetry (DSC) to analyze mixtures of CuO and Al2O3 in two proportions, with 0.32 and 0.75 mol fraction of CuO, up to 1573 K in air and in a protective atmosphere of N2 [30]. Arjmand et al. measured the standard enthalpy of formation of CuAl2O4 by DSC-TGA in a reduction experiment with CO at 1173 K [11]. Chen and Zhao investigated the phase equilibria of the Cu2O–Al2O3 system at metallic copper saturation experimentally by applying high-temperature equilibration, quenching, and electron probe microanalysis (EPMA). A new set of experimental data was reported from 1473 to 1723 K [31]. Hellstén et al. [32] investigated phase equilibria in the Cu–O–Al2O3 system from 1373 to 1673 K in air by the equilibration/quenching method with a subsequent elemental analysis of the equilibrium phases by EPMA. The Al2O3–Cu2O projected isobaric section was developed [32], where the oxide liquid phase with significantly smaller alumina content was depicted in comparison with the previous studies.
Crystallographic information of copper oxides and copper aluminates is compiled in Table 1.
Nevertheless, the available thermodynamic data are discrete and incomplete. Moreover, there is a discrepancy among the reported experimental data. The purpose of the present study is to determine experimentally a new set of data including the thermodynamic stability of the solid phases of the Cu–O–Al2O3 system and the phase transition enthalpies and temperatures occurring on the Al2O3–CuAlO2 and the Al2O3–CuAl2O4 tie-lines and to critically discuss them among the previously reported data.
Experimental
Materials and synthesis
The cuprous oxide (Cu2O) was synthesized by oxidizing cathode copper (0.99996 mass fraction, Boliden Harjavalta Oy, Finland). The high-purity copper was annealed in air above 1299 K for 120 h and quenched. The formation of the cuprous oxide was confirmed by EPMA and its purity was estimated to be at least 0.9999 mass fraction.
High-purity powders of alumina (Al2O3, 0.9999 mass fraction, Sigma-Aldrich, USA) and cupric oxide (CuO, 0.99995, Alfa-Aesar, Germany) were used as reactants for synthesis of CuAl2O4. The fine alumina powder was heat treated at 1373 K for 2 h to remove any moisture and volatile impurities. Then, Al2O3 and CuO were thoroughly mixed in an agate mortar with molar ratio of 2:1 and pressed using 0.25 GPa into a cylindrical pellet (15 mm in diameter and approximately 5 mm high). This sample was subjected to three heat treatment cycles (24 h each) in air at 1338 K with intermediate grinding and pelletizing stages. The obtained sample had a brownish color. Conducted X-ray powder diffraction (XRPD) analysis and scanning electron microscopy with energy-dispersive spectroscopy (SEM-EDS) analysis of the polished section confirmed that the synthesized sample was CuAl2O4 with alumina saturation. Based on the semiquantitative XRPD analysis, the sample phase composition was CuAl2O4/Al2O3 = 62/38 wt% (0.9/1 mol ratio).
Direct synthesis of CuAlO2 from its component oxides was unsuccessful. Despite an excessive amount of alumina, a small fraction of Cu2O was remaining unreacted even after multiple grinding and heat treatment cycles. Thus, CuAlO2 was synthesized in a different way from the obtained sample of CuAl2O4 with alumina saturation. The sample was decomposed at 1473 K for 14 h and cooled down to room temperature with 4 K min−1 cooling rate in a protective atmosphere of argon. The obtained sample had a light-bluish color. XRPD and SEM-EDS examinations revealed the formation of CuAlO2 with alumina saturation. Based on the semiquantitative XRPD analysis, the sample phase composition was CuAlO2/Al2O3 = 36/64 wt% (0.7/1.5 mol ratio).
In both synthesized aluminate samples, the molar ratios of the phases present deviated from the theoretical ones calculated from the amount of the reactants used and considering reaction paths (1) and (2), i.e., 1:1 and 1:1.5, respectively, probably due to the observed reactions of the samples with alumina crucibles used as a substrate during the annealings.
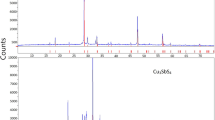
Nevertheless, both samples were qualitatively pure and their XRPD patterns are depicted in Fig. 1. Also, micrographs of their polished sections (Figs. S1 and S2) with respective elemental compositions of the ternary compounds (Tables S1 and S2) obtained by the SEM-EDS analysis are presented in Electronic Supplementary Material (ESM). Two phase assemblages, i.e., CuAl2O4–Al2O3 and CuAlO2–Al2O3, can be seen in Figs. S1 and S2, respectively. The SEM-EDS analysis was applied as a supplementary tool in order to confirm the XRPD findings concerning the formation of copper aluminates. The EDS analysis of such samples produced through the ceramic route is associated with difficulties related to a high porosity.

XRPD patterns of the synthesized samples. a CuAl2O4 at Al2O3 saturation; b CuAlO2 at Al2O3 saturation; and the standard peaks marked: (circle) Al2O3; (diamond) CuAl2O4; (triangle) CuAlO2
Analysis
In the XRPD analysis, specimens in a fine powder form were examined by a PANalytical diffractometer (X’Pert Pro Powder, Almelo, Netherlands) equipped with CuKα radiation with a step size of 0.007° over a 2θ range of 10–70° at room temperature. In the phase composition identification of the samples, Pan-Inorganic Crystal Structure Database (ICSD) was used. Standard patterns with the following ICSD collection codes were used for the respective substances: CuO—069757, Cu2O—063281, Al2O3—088027, CuAl2O4—172131, and CuAlO2—31701.
For the SEM-EDS analysis, polished sections of the specimens were carbon-coated and examined by a LEO 1450 (Carl Zeiss Microscopy GmbH, Jena, Germany) scanning electron microscope with a Link Inca X-Sight 7366 energy EDS analyzer (Oxford Instruments plc, Abingdon, Oxfordshire, UK). Olivine, pure aluminum, and cuprous oxide (Cu2O) were employed as standards for O, Al, and Cu, respectively. Kα spectral lines were utilized for each element. The accelerating voltage used was 15 kV.
In the EPMA, polished and carbon-coated specimens were analyzed by CAMECA SX100 (Cameca SAS, France). Pure metallic copper and hematite were employed as standards for Cu and O, respectively. Kα spectral lines were utilized for each element. The accelerating voltage of 15 kV and beam current of 40 nA were used in the analysis.
Arrangement of EMF setups for the copper oxides
Reversible EMF values of the oxygen concentration galvanic cells based on the solid oxide electrolytes:
were measured within the temperature ranges of 1003–1322 and 907–1232 K, respectively, for determination of the standard Gibbs energy of formation of Cu2O and CuO.
Two independent high-temperature experimental setups (vertical and horizontal) were employed for the determination of thermodynamic stabilities of the copper oxides. The schematics of the setups are depicted in Figs. 2 and 3 for the vertical and horizontal designs, respectively. High-purity oxygen and argon gases utilized in the EMF experiments were supplied by AGA, Finland (both were 0.99999 volume fraction). Prior to use, argon was purified from its original purity by passing through chopped copper foil at 673 K and titanium turnings at 1173 K.

Schematics of the vertical tube furnace: 1, vertical tube furnace; 2, mullite work tube; 3, CSZ crucible; 4, upper alumina tube; 5, CSZ tube; 6, lower alumina tube; 7, Pt lead wire (test side); 8, test electrode; 9, Pt lead wire (reference side); 10, thermocouple

Schematics of the horizontal tube furnace (the arrangement of the galvanic cell and the reference electrode is enlarged): 1, horizontal tube furnace; 2, fused silica work tube; 3, argon gas inlet; 4, argon gas outlet; 5, oxygen gas inlet; 6, oxygen gas outlet; 7, steel springs; 8, alumina sheath; 9, left side thermocouple; 10, alumina cell holder; 11, alumina crucible; 12, piece of alumina tube; 13, Pt lead wire (test side); 14, test electrode; 15, Pt lead wire (reference side); 16, YSZ tube; 17, alumina tube; 18, reducing tube-to-tube joint; 19, steel tube; 20, front and back ferrules; 21, plug; 22, cross joint; 23, right side thermocouple
The high-temperature apparatus employed was based on a vertical Lenton resistance tube furnace type CSC 12/−−/600 V. The electrochemical cell was assembled inside a mullite work tube. The reference electrode compartment was arranged in a long calcia-stabilized zirconia (CSZ) tube with a closed flat end. A platinum contact in a shape of a coil was pressed to the inner surface of the bottom of the CSZ tube by an alumina tube. Pure oxygen gas was passing through this alumina tube and flushing the inner surface of the CSZ tube. Also, a calibrated S-type thermocouple was inserted into the alumina tube in such a way that its junction was located right next to the bottom of the solid electrolyte tube.
Prior to the experiments, the temperature profile of the furnace along its vertical axis was measured with two identical S-type thermocouples located right below and above the bottom of the solid electrolyte tube to determine the hot zone of the furnace with negligible temperature gradient. The temperature difference between the two thermocouples was less than 1 K within the whole experimental temperature range. Then, the actual experiments were conducted without upper thermocouple.
The test electrode was arranged in a CSZ crucible. A powder mixture of either Cu + Cu2O (cell A) or Cu2O + CuO (cell B) was pressed into a 2–3-mm thick pellet. The utilized high-purity Cu powder (0.99995 mass fraction, Alfa-Aesar, Germany) was reduced in a gas mixture of 10 % H2–90 % Ar (AGA, Finland) at 973 K for 1 h, prior the electrode preparation. The pressed pellet with a platinum contact was placed into the solid electrolyte crucible. And, the crucible was placed on the CSZ tube (see Fig. 2), while the closed-end upper alumina tube pressed the whole assembly together providing perfect contacts on the interfaces. Both contacted CSZ surfaces were polished beforehand. The test electrode compartment was flushed with purified argon providing nearly static protective atmosphere.
The horizontal setup adapted for the EMF measurements was based on a Lenton resistance tube furnace-type CSC 12/−−/600H. It is illustrated in Fig. 3. This furnace was equipped with a fused silica work tube, where the electrochemical cell was assembled. The reference electrode was arranged inside a long yttria-stabilized zirconia (YSZ) tube by passing a pure oxygen gas through an inserted alumina tube and flushing the inner surface of the YSZ tube. Also, this alumina tube pressed a tip of a Pt wire in the form of a coil to the inner surface of the bottom of the solid electrolyte tube, forming an electrical contact. The arrangement of the gas-tight oxygen inlet and outlet trains is illustrated in Fig. 3.
The test electrode pellets used in this setup were identical to those used in the vertical setup. The pellet with a Pt contact was pressed to the other side of the solid electrolyte tube. An alumina crucible and an alumina tube were utilized to support the test electrode pellet in the center of the alumina cell holder. The spring-loaded solid electrolyte tube pressed the pellet to the Pt contact, providing reliable contacts. Two S-type thermocouples were used to dispose the galvanic cell into the isothermal zone. One thermocouple was located in the alumina cell holder; the other one was inside the YSZ tube (Fig. 3). The temperature difference less than 1 K between the thermocouples was achieved by adjusting the position of the galvanic cell inside the furnace. The test electrode was protected by nearly static atmosphere of purified argon gas.
Redesigned EMF setups for the copper aluminates investigation
For determination of thermodynamic stability of the copper aluminates, reversible EMF values of the solid-state galvanic cells based on magnesia-stabilized zirconia (MSZ) electrolyte:
were measured within the temperature ranges of 1051–1265 and 1074–1262 K, respectively.
The pelletized test electrodes for the galvanic cells (C) and (D) were comprised of three phases, Cu + CuAlO2 + Al2O3 and CuAl2O4 + CuAlO2 + Al2O3, respectively. The design of galvanic cells employed for copper oxides measurements failed in the case of copper aluminates. Hence, a rather open original arrangement of the test electrodes (Figs. 2 and 3) was improved by an insulation with an alumina cement. Due to design-related reasons, the gaseous reference electrode was replaced by either Cu + Cu2O (cell C) or Cu2O + CuO (cell D) mixtures and the reference electrode compartment was sealed in a similar manner with an alumina cement. The redesigned electrochemical cells are depicted in Figs. 4 and 5. A near-static protective atmosphere of the purified argon gas was provided in the work tube.

Schematics of the redesigned vertical tube furnace: 1, vertical tube furnace; 2, mullite work tube; 3, MSZ tubular crucible; 4, upper alumina tube; 5, reference electrode; 6, test electrode; 7, alumina crucibles; 8, lower alumina tube; 9, Pt lead wires; 10, alumina cement; 11, thermocouple

Schematics of the redesigned horizontal tube furnace (an enlarged arrangement of the galvanic cell): 1, alumina cell holder; 2, alumina sheath; 3, left side thermocouple; 4, alumina crucible; 5, reference electrode; 6, test electrode; 7, MSZ tubular crucible; 8, alumina cement; 9, alumina tube; 10, right side thermocouple; 11, Pt lead wires
Several types of stabilized zirconia solid electrolytes were utilized in this study. In the arrangement depicted in Fig. 2, a tube (17 × 20 × 610 mm) and a crucible (17 × 20 × 40 mm) made of calcia-stabilized zirconia (3.5 wt% CaO) were supplied by Corning, Zircoa Products (Solon, USA). In the horizontal apparatus shown in Fig. 3, the utilized yttria-stabilized zirconia (8.5 wt% Y2O3) tube (5 × 8 × 800 mm) with a roundish end was supplied by Friatec (Germany). In the modified arrangements of the electrochemical cells (Figs. 4 and 5), tubular solid electrolytes (9 × 13 × 50 mm) with a flat end made of magnesia-stabilized zirconia (15 mol% MgO) were supplied by Yamari (Japan). The choice of the electrolyte types was related to quality, availability, geometrical shapes, and dimensions.
Polarity of oxygen concentration cells (A), (B), and (C) is obvious, but for cell (D), a positive EMF was measured only at higher temperatures, while at lower ones, it changed its sign. However, in the discussions and in the calculations, the left-hand side electrode is called and considered as a test electrode in all the cases.
For the measurement of the EMF values produced by the galvanic cells, the identical Pt lead wires (Johnson-Matthey Noble Metals, UK) were connected to a Keithley 6517B electrometer with high input impedance of 2 × 1014 Ω. It enabled conducting the EMF measurements in an open circuit. The galvanic cell temperature measurements were conducted with calibrated S-type Pt-Pt/Rh thermocouples (Johnson-Matthey Noble Metals, UK) connected to a Keithley 2010 DMM multimeters. A Pt100 resistance thermometer (SKS-Group, Finland, tolerance class B 1/10) connected to a Keithley 2000 DMM multimeter was utilized as a cold junction compensation. The experimental temperature and the EMF values were simultaneously recorded by a computer at a rate of one datapoint per 5 s. Also, for galvanic cells (A) and (B), where the gaseous reference electrode was employed, the ambient pressure was measured with a Vaisala PTU300 (Finland) (pressure, humidity, and temperature transmitter) and recorded in order to compensate its natural fluctuation in the calculations of the thermodynamic properties.
In selected experiments, the gas atmosphere on the furnace outlet was analyzed with an oxygen probe, Cambridge Sensotec Rapidox 2100 (UK), to assure that the protective atmosphere of argon gas was sufficiently purified.
DSC-TGA techniques
A combined use of DSC-TGA techniques constitute the most convenient method for characterizing phase transition properties. In this study, thermal analyses of the synthesized samples were done by the DSC-TGA techniques using a NETZSCH STA 449 F1 Jupiter (Germany)–Simultaneous DSC-TGA. The calorimeter was calibrated with the melting temperatures and enthalpies of fusion for high-purity tin, indium, bismuth, zinc, aluminum, and gold. The average measurement accuracies of temperatures and enthalpies of fusion were determined to be ±1 K and ±1.14 %, respectively.
The calorimetric measurements were performed with the synthesized samples of copper aluminates saturated with alumina. Weight loss and heat flow during the linear heating and cooling (10 K min−1) in all runs were measured simultaneously. Al2O3 crucibles of the same mass were used for both the base line (blank run) and thermal analysis of the samples. Before the start of each experiment, the chamber was evacuated and then backfilled with pure N2 supplied by AGA, Finland (0.99999 volume fraction), three times to remove traces of reactive gases. The runs were performed either in a protective atmosphere of pure N2 or in a N2–O2 mixture (around 0.143 atm of oxygen partial pressure) for delafossite and spinel phases, respectively.
Results and discussions
Determination of the standard Gibbs energies of formation of solid phases in the Cu–O–Al2O3 system
According to the Gibbs phase rule for the binary Cu–O system, three condensed phases coexist with the gas phase in an invariant point at only one temperature and oxygen partial pressure, and the number of condensed phases coexisting with the gas phase becomes four in an invariant equilibrium if the whole ternary Cu–O–Al2O3 system is considered. Meanwhile, in the present EMF measurements, the monovariant equilibria were investigated, i.e., for the binary Cu–O system, two condensed phases, either Cu + Cu2O (in cell (A)) or Cu2O + CuO (in cell (B)), were equilibrated in isothermal conditions and the equilibrium oxygen partial pressure was measured. Thus, at a given temperature, three phases (two solids and gas) coexist at only one equilibrium oxygen partial pressure which was measured directly with the electrochemical cells. In a similar way, for the Cu–O–Al2O3 system, in cells (C) and (D), monovariant equilibria (three solids and gas) were investigated. By equilibrating the galvanic cells stepwise at different temperatures, the equilibrium oxygen partial pressure was measured as a function of temperature for the equilibria (3–6):
Eqs. (3–6) represent the reaction equilibria attained at the test electrodes of the galvanic cells (A–D), respectively.
The equilibrium oxygen partial pressure (or oxygen activity) at the test electrode was calculated from the experimentally measured values by using the Nernst equation:
where E and T stand for the experimentally measured EMF (V) and the temperature (K), R and F are the universal gas constant (8.314472 J mol−1 K−1) [37] and the Faraday constant (96,485.3399 C mol−1) [37], and a O2(r) and a O2(t) are the oxygen activities at the reference and the test electrodes, respectively.
For cells (A) and (B), the oxygen activity at the reference electrode was close to 1 within the entire measurements, since high-purity O2 was utilized at the ambient pressure. However, due to the natural fluctuation of ambient pressure and an insignificant deviation from the standard state, Eq. (8) was used for the respective correction:
where P is the ambient pressure measured during the entire experiment and recorded for every measurement point, and P° is the standard pressure (0.1 MPa) [37].
Ultimately, the standard Gibbs energies of reactions (3–6) were calculated from Eq. (9), where K stands for the equilibrium constant calculated as a ratio of activities of the reaction products to the reactants raised to the power of the stoichiometric number. Activities of the condensed species in reactions (3–6) were assumed to be 1, since a significant deviation from the stoichiometry of the compounds was neither reported nor observed. Hence, the equilibrium constant can be calculated from the activity of oxygen at the test electrode calculated from Eq. (7).
Before the calculations of the thermodynamic properties, raw data were analyzed and processed. For the EMF vs. time plot recorded, averages of 200 data points (1000 s) were calculated for every equilibrium. The original signal was treated with median filter beforehand for discrimination of the signal noise. Respective averages of the temperature (and the pressure for cells (A) and (B)) were calculated for the same period of time for every measurement point. These average values calculated for every measurement point with respective standard deviations of EMF are given in Tables S3–S6 (ESM) for galvanic cells (A)–(D), respectively. The numbers of experimental galvanic cells used with their life time are also provided in Tables S3–S6. The standard Gibbs energies of reactions (3–6) were calculated for every experimental point measured and then the best linear fit was calculated.
The standard Gibbs energies of reactions (3) and (4) are depicted in Figs. 6 and 7, respectively. The round symbols represent the individual measurement points obtained with cells (A) and (B). The calculated best linear fits with determination coefficient R2 of 0.9947 and 0.9882, respectively, are expressed in Eqs. (10) and (11) with their respective standard uncertainties and compared with the selected previous studies in Figs. 6 and 7:

The standard Gibbs energy of reaction (3) determined in this study and compared with the previous works

The standard Gibbs energy of reaction (4) determined in this study and compared with the previous works
The monovariant equilibria of Cu + Cu2O + gas and Cu2O + CuO + gas are shown in Fig. 8 with the same measurement points depicted in Figs. 6 and 7, expressed as P O2 vs. T. They are superimposed on the potential P O2-T phase diagram assessed by Shishin and Decterov [15] for comparison.

The experimentally measured oxygen partial pressure as a function of temperature of Cu–Cu2O and Cu2O–CuO equilibria depicted on an assessed Cu–O phase diagram [15]
Figures 6, 7, and 8 show an excellent agreement with this study and the research works published previously [4, 15, 23, 26, 38]. These measurements (cells (A) and (B)) are new sets of experimental data, which confirm reliability of the thermodynamic properties determined for cupric and cuprous oxides previously. Furthermore, the measurements also confirm the reliability of the experimental setups, devices, and the entire technique applied in this study, absence of a significant systematic error.
The standard Gibbs energies of reactions (5) and (6) are plotted in Figs. 9 and 10 as a function of temperature. The round labels present the individual measurement points obtained with cells (C) and (D). The calculated best linear fits are expressed in Eqs. (12) and (13) with their respective standard uncertainties and compared with the previous studies in Figs. 9 and 10. The obtained R2 determination coefficients are 0.9742 and 0.9901, respectively.

The standard Gibbs energy of reaction (5) determined in this study and compared with the previous works

The standard Gibbs energy of reaction (6) determined in this study and compared with the previous works
Figure 9 shows a scatter among the results of the previous studies for the Gibbs energy of reaction (5). The ∆r G° values measured in the previous studies at 1150 K do not differ from the present one for more than 4.2 kJ mol−1, but the discrepancy increases at lower and higher temperature due to the differences in the slope. Therefore, the extrapolation can cause a significant error and should be avoided.
Figure 10 shows that the Gibbs energy of reaction (6) measured in this study is in a good agreement with the results obtained by Jacob and Alcock [4]. The results reported by Gadalla and White were measured only in a narrow range of temperature, and therefore the slope is not reliable. However, if ∆r G° measured in this study are extrapolated to the higher temperature, they agree well with [23]. The results reported by Slobodyanyuk et al. [26] are about two times more negative and probably less accurate, due to an unknown systematic error.
From Eqs. (10–13), the standard Gibbs energies of formation of copper oxides from the elements and of copper aluminates from their component oxides can be calculated by applying Eqs. (14–17).
E C and E D stand for the EMF of cells (C) and (D), respectively. Thus, the standard Gibbs energies of formation can be expressed by Eqs. (18–21) with their respective standard uncertainties:
The standard Gibbs energies of formation of Cu2O and CuO are in a good agreement with the previous studies [4, 15, 23, 26, 38] as well as with the values reported in the thermodynamic compilations [39].
The obtained stability functions of copper aluminates (Eqs. (20) and (21)) are compared with results of the previous studies in Tables 2 and 3.
Also, Navrotsky and Kleppa [24] measured calorimetrically the standard enthalpy of formation of CuAl2O4 from its component oxides as 21.63 ± 0.79 kJ mol−1 and Arjmand et al. [11] measured the standard enthalpy of formation of CuAl2O4 from the elements as −1824.4 ± 4.1 kJ mol−1.
The standard Gibbs energy of formation of CuAl2O4 from its component oxides determined in the previous electrochemical studies [4, 26] and in the present work was derived from several individual phase equilibria (see Eq. (17)) measured with respective galvanic cells. For this reason, the possible uncertainties of the individual measurements were accumulated and a certain discrepancy can be seen among the results reported in Table 3. For instance, at 1200 K, the ∆f(ox) G°CuAl2O4 values obtained in the present work agree well with the results obtained by Jacob and Alcock [4], but there is a large discrepancy at lower temperatures, since the slopes are opposite each other. Therefore, an extrapolation within a wide temperature range can cause a large error and should be avoided. Meanwhile, the decomposition temperature of CuAl2O4 into its component oxides was derived from the indirect measurements [4] by extrapolation of the high temperature data up to 885 K. Thus, a direct experiment on the decomposition of CuAl2O4 was conducted in order to solve the discrepancy.
The decomposition test of CuAl2O4
Several samples of CuAl2O4 at Al2O3 saturation pressed into pellets were annealed in air for 120 h at temperatures below 885 K: 773, 823, and 853 K. The polished sections of the annealed samples were analyzed by SEM. From the micrographs, no free CuO was observed; however, due to a small particles size (a few μm) of the starting biphasic mixture and high sample porosity, the results of EDS measurements cannot be considered as reliable ones. Therefore, the annealed samples were additionally analyzed by XRPD. Figure 11 shows patterns of the initial spinel at alumina saturation and the samples after annealing. No changes of the phase composition can be indicated.

The XRPD patterns of the CuAl2O4–Al2O3 samples annealed at different temperatures; the standard peaks marked: (circle) Al2O3; (diamond) CuAl2O4
In Fig. 11, the decomposition was not observed due to either lower decomposition temperature or extremely slow kinetics of this process. Previously, CuAl2O4 was annealed at 873 K in air within 26 h in an independent study and no decomposition was reported [35].
Determination of phase transition enthalpies and temperatures of CuAlO2 and CuAl2O4
In the present work, the DSC-TGA measurements were conducted for the synthesized phases of CuAl2O4–Al2O3 and CuAlO2–Al2O3 in alumina crucibles. The DSC-TGA curves obtained are presented in Figs. 12 and 13. The enthalpies of phase transitions were calculated as integrals between onset and end temperatures of the peaks. The temperatures of the measured phase transitions were obtained from onsets on the heating curves; however, cooling curves were also considered in discussions.

The DSC-TGA curves as a function of temperature for the CuAl2O4–Al2O3 in N2–O2 gas atmosphere with a linear heating/cooling rate of 10 K min−1

The DSC-TGA curves as a function of temperature for the CuAlO2–Al2O3 in N2 gas atmosphere with a linear heating/cooling rate of 10 K min−1
The first run (Fig. 12) for the CuAl2O4 saturated with Al2O3 was performed at 0.143 atm of oxygen partial pressure (N2–O2 mixture). The furnace was heated to 298 K and kept at an isothermal condition for few minutes. Then, the furnace was heated to 1573 K and cooled to 1073 K at a linear heating/cooling rate of 10 K min−1. The second run (Fig. 13) for the CuAlO2 saturated with Al2O3 was conducted in a protective atmosphere (pure N2). The furnace was first heated to 323 K and kept at isothermal conditions for few minutes. Then, the furnace was heated to 1633 K and cooled to 1413 K at a linear heating/cooling rate of 10 K min−1.
Two endothermic peaks on the heating DSC curve can be seen in Fig. 12, where spinel was examined in an oxidizing atmosphere (P O2 = 0.143 atm). The first peak indicates a decomposition of spinel phase into delafossite and alumina with evolution of oxygen gas, and the TG curve shows respective weigh loss of 2.53 wt.% (the theoretical weight loss for the mixture of 62 wt% CuAl2O4 and 38 wt.% Al2O3 is 2.73 wt.% according to reaction (2)). The onset temperature of the first peak (1416.7 K) was determined to be the temperature of the decomposition. Enthalpy of the reaction was calculated as −3.64 kJ mol−1, taking into account the initial phase composition (the fraction of alumina did not contribute and was ignored).
The second peak indicates an incongruent melting of delafossite phase into oxide liquid and solid alumina with noticeable oxygen capture from the gas phase by the melt toward its equilibrium composition. The onset temperature of the second peak (1505.4 K) was determined to be the temperature of the melting. Hence, delafossite phase is stable within this narrow temperature range from 1416.7 to 1505.4 K at 0.143 atm of oxygen partial pressure. The enthalpy of delafossite melting was calculated, assuming that decomposition of spinel was complete and composition of the specimen before melting was 41.85 wt.% CuAlO2 and 55.42 wt.% Al2O3 (total is 100, if 2.73 wt.% of lost O2 is considered). Also, the alumina fraction present was considered as inert material in a similar way as in the previous reaction and a thermal effect of the oxygen capture by the melt was neglected. Therefore, the enthalpy of the reaction was calculated as −1.80 kJ mol−1. The enthalpies of reactions were calculated in the way described above, because certain mass changes occurred during the run and the initial mass of the sample was used in measurements of enthalpies.
On the cooling curve, an exothermic crystallization of stoichiometric delafossite occurred with respective slight weight loss. The formation of spinel phase on the cooling curve did not occur, probably due to kinetic reasons.
One endothermic peak on the heating DSC curve can be seen in Fig. 13, where delafossite was examined in a protective atmosphere of pure N2. This peak corresponds to incongruent melting of delafossite phase into oxide liquid and solid alumina. From the TGA curve, it can be seen that no oxygen was captured since the experiment was conducted in pure N2. Instead, a negligible weight loss probably associated with a vaporization can be seen. The onset temperature of the melting was measured at 1555.9 K. The enthalpy of this reaction was calculated as −2.14 kJ mol−1 (the initial specimen composition was 36 wt.% CuAlO2 and 64 wt.% Al2O3 and it was used in the calculation), considering the alumina fraction as an inert material.
On the heating curve, no other peaks can be observed. On the cooling curve, an exothermic peak indicates a crystallization of delafossite phase.
The enthalpies of discussed reactions as well as respective temperatures are summarized in Table 4 and compared with the literature data.
The enthalpy of melting of delafossite measured at oxygen partial pressure of 0.143 atm and in pure N2 slightly differ from each other, probably due to an exothermic process of O2 capturing by the melt, taking place in former case, and simultaneously with the actual melting.
In addition to these two DSC-TGA runs discussed, a preliminary run focused on decomposition of spinel phase in a protective atmosphere of pure N2 was conducted with the same linear heating/cooling rate of 10 K min−1. This experiment revealed that CuAl2O4 is not stable in non-oxidizing conditions; it decomposes according to reaction path described in Eq. (2) with the respective weight loss associated with O2 release. According to the TGA curve in Fig. S3 (ESM), the decomposition started at about 1200 K and completed at about 1400 K. This process occurred gradually within a wide (about 200 K) temperature range, and it starts at much lower temperatures than in the oxidizing conditions (see Fig. 12). The DSC curve was not considered. Such a gradual process can be explained by kinetic limitations at lower temperatures. These observations are in a good agreement with the EMF data. The equilibrium oxygen partial pressures of reaction (6) with respective ∆r(6) G° values calculated for several selected temperatures are presented in Table 5.
Obviously, the equilibrium P O2 exceeds the small P O2 (traces of oxygen) of N2 gas supplied with 0.99999 purity even at 1000 K. Therefore, the decomposition proceeds with oxygen loss.
Conclusions
Thermodynamic properties of Cu2O and CuO were experimentally measured with the solid electrolyte EMF method, employing the pure oxygen gas as the reference electrode. The same experimental EMF setups were accordingly redesigned for determination of the standard Gibbs energies of formation of CuAlO2 and CuAl2O4. The comparison of the measured stability functions with the literature data revealed a rather large discrepancy, mainly observed on the slope among this work and the previous studies. A direct experiment focused on the low temperature decomposition of spinel (CuAl2O4) into its component oxides revealed that it does not occur in the temperature range of interest. Combined DSC-TGA techniques were employed to investigate the phase transitions occurring on the CuO–Al2O3 and the Cu2O–Al2O3 tie-lines. Consequently, the enthalpies of the reactions with reaction temperatures were determined in various gas atmospheres. All the results obtained are discussed in the framework of the available literature data reported previously.
References
Schlesinger ME, King MJ, Sole KC, Davenport WGI (2011) Extractive metallurgy of copper (chapter 18), fifth edn. Elsevier, Oxford
Malfliet A, Lotfian S, Scheunis L, Petkov V, Pandelaers L, Jones PT, Blanpain B (2014) Degradation mechanisms and use of refractory linings in copper production processes: a critical review. J Eur Ceram Soc 34(3):849–876
Rigby GR, Hamilton B (1961) A study of basic brick from copper anode furnaces. J Am Ceram Soc 44:201–205
Jacob KT, Alcock CB (1975) Thermodynamics of CuAlO2 and CuAl2O4 and phase equilibria in the system Cu2O-CuO-Al2O3. J Am Ceram Soc 58(5–6):192–195
Fujimura T, Tanaka S-I (1998) In-situ high temperature X-ray diffraction study of Cu/Al2O3 interface reactions. Acta Mater 46(9):3057–3061
YI S, Trumble KP, Gaskell DR (1999) Thermodynamic analysis of aluminate stability in the eutectic bonding of copper with alumina. Acta Mater 47(11):3221–3226
Seager CW, Kokini K, Trumble K, Krane MJM (2002) The influence of CuAlO2 on the strength of eutectically bonded Cu/Al2O3 interfaces. Scr Mater 46:395–400
Silvain JF, Bobet JL, Heintz JM (2002) Electroless deposition of copper onto alumina sub-micronic powders and sintering. Compos A: Appl Sci Manuf 33A(10):1387–1390
Tahir D, Tougaard S (2012) Electronic and optical properties of Cu, CuO and Cu2O studied by electron spectroscopy. J Phys Condens Matter 24(17):175002/1–175002/8
Robertson J, Peacock PW, Towler MD, Needs R (2002) Electronic structure of p-type conducting transparent oxides. Thin Solid Films 411(1):96–100
Arjmand M, Knee CS, Leion H, Mattisson T (2015) Standard enthalpy of formation of CuAl2O4 revisited. Chem Eng Commun 202(5):694–697
Hallstedt B, Risold D, Gauckler LJ (1994) Thermodynamic assessment of the copper-oxygen system. J Phase Equilib 15(5):483–499
Hallstedt B, Gauckler LJ (2003) Revision of the thermodynamic descriptions of the Cu-O, Ag-O, Ag-Cu-O, Bi-Sr-O, Bi-Ca-O, Bi-Cu-O, Sr-Cu-O, Ca-Cu-O and Sr-Ca-Cu-O systems. Calphad 27(2):177–191
Clavaguera-Mora MT, Touron JL, Rodriguez-Viejo J, Clavaguera N (2004) Thermodynamic description of the Cu-O system. J Alloys Compd 377(1–2):8–16
Shishin D, Decterov SA (2012) Critical assessment and thermodynamic modeling of the Cu-O and Cu-O-S systems. Calphad 38:59–70
Roberts HS, Smith FH (1921) The system copper: cupric oxide: oxygen. J Am Ceram Soc 43:1061–1070
Gadalla AMM, Ford WF, White J (1963) Equilibrium relations in the system CuO-Cu2O-SiO2. Brit Ceram Trans J 62(1):45–66
Fitzner K, Moser Z (1979) Activity of oxygen in dilute liquid copper-oxygen alloys. Metals Technol (London) 6(7):273–275
Boudene A, Hack K, Mohammad A, Neuschutz D, Zimmermann E (1992) Experimental investigation and thermochemical assessment of the system copper-oxygen. Zeitschrift fuer Metallkunde 83(9):663–668
Kosenko AV, Emel’chenko GA (2001) Equilibrium phase relationships in the system Cu-O under high oxygen pressure. J Phase Equilib 22(1):12–19
Schmalzried H (1960) Measurement of the free enthalpy of reaction in the formation of spinel phases from the single oxides by aid of solid galvanic couples. Zeitschrift fuer Physikalische Chemie (Muenchen, Germany) 25:178–192
Misra SK, Chaklader ACD (1963) The system copper oxide–alumina. J Am Ceram Soc 46(10):509
Gadalla AMM, White J (1964) Equilibrium relation in the system Cu2O-CuO-Al2O3. Brit Ceram Trans J 63(1):39–62
Navrotsky A, Kleppa OJ (1968) Thermodynamics of formation of simple spinels. J Inorg Nucl Chem 30(2):479–498
Zalazinskii AG, Balakirev VF, Chebotaev NM, Chufarov GI (1969) Thermodynamic analysis of the reduction, dissociation, and formation of copper aluminate, copper chromate(III), and copper ferrate(II) from the free elements and oxides. Zhurnal Neorganicheskoi Khimii 14(3):624–626
Slobodyanyuk AA, Tret’yakov YD, Bessonov AF (1971) Thermodynamic stability of copper silicates and aluminates studied by emf. with a solid electrolyte. Zhurnal Fizicheskoi Khimii 45(7):1871–1872
Tsuchida T, Furuichi R, Sukegawa T, Furudate M, Ishii T (1984) Thermoanalytical study on the reaction of the CuO-Al2O3 (η, γ and α) systems. Thermochim Acta 78(1–3):71–80
Trumble KP (1992) Thermodynamic analysis of aluminate formation at Fe/Al2O3 and Cu/Al2O3 interfaces. Acta Metall Mater 40(Suppl):S105–S110
Trumble KP (1999) Prediction of a critical temperature for aluminate formation in alumina/copper-oxygen eutectic bonding. J Am Ceram Soc 82(10):2919–2920
Guedes M, Ferreira JMF, Ferro AC (2008) A study on CuO-Al2O3 reaction paths. Adv Powder Metall Part Mater 5/1–5/13
Chen M, Zhao B (2013) Phase equilibrium studies of "Cu2O"-SiO2-Al2O3 system in equilibrium with metallic copper. J Am Ceram Soc 96(11):3631–3636
Hellstén N, Hamuyuni J, Taskinen P (2016) High-temperature phase equilibria of Cu-O-Al2O3 system in air. Can Metall Q 55(2):226–233
Åsbrink S, Waskowska A (1991) CuO: X-ray single-crystal structure determination at 196 K and room temperature. J Phys Condens Matter 3(42):8173–8180
Restori R, Schwarzenbach D (1986) Charge density in cuprite, Cu2O. Acta Crystallogr Sect B: Struct Sci B42(3):201–208
O’Neill HSC, James M, Dollase WA, Redfern SAT (2005) Temperature dependence of the cation distribution in CuAl2O4 spinel. Eur J Mineral 17(4):581–586
Ishiguro T, Kitazawa A, Mizutani N, Kato M (1981) Single-crystal growth and crystal structure refinement of CuAlO2. J Solid State Chem 40(2):170–174
Cohen ER, Cvitaš T, Frey JG, Holmström B, Kuchitsu K, Marquardt R, Mills I, Pavese F, Quack M, Stohner J, Strauss HL, Takami M, Thor AJ (2007) Quantities, units, and symbols in physical chemistry IUPAC green book, 3rd edn. IUPAC and RCS Publishing, Cambridge
Holmes RD, O’Neill HSC, Arculus RJ (1986) Standard Gibbs free energy of formation for Cu2O, NiO, CoO, and FexO: high resolution electrochemical measurements using zirconia solid electrolytes from 900 to 1400 K. Geochim Cosmochim Acta 50(11):2439–2452
Barin I (1989) Thermodynamical data of pure substances, part I, VCH Verlagsgesellschaft, Weinheim. VCH Publishers, New York
Acknowledgments
This work was financially supported by the Finnish Metals Producers Fund and the Academy of Finland.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(PDF 264 kb)
Rights and permissions
About this article

Cite this article
Sukhomlinov, D., Tesfaye, F., Hellstén, N. et al. Thermodynamic stability of solid phases in the system Cu–O–Al2O3 by means of the EMF and DSC-TGA techniques. J Solid State Electrochem 22, 959–972 (2018). https://doi.org/10.1007/s10008-016-3430-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-016-3430-1