
Abstract
Carbon-supported PtCu and PtSn (both with an atomic ratio of 3:1) nanoparticles were prepared by reducing Pt, Sn, and Cu precursors via refluxing in ethanol. They were characterized using energy-dispersive X-ray spectroscopy (EDX), X-ray diffraction (XRD), and transmission electron microscopy (TEM). X-ray diffraction indicated that the lattice parameter of Pt increases with the addition of Sn and decreases with the addition of Cu, indicating that both PtCu and PtSn are solid solutions. EDX analysis revealed that the compositions of these materials are close to their nominal compositions, while TEM showed that both materials presented a homogeneous particle distribution on the carbon support and low particle agglomeration. Electrochemical experiments indicated that all of the materials are electrochemically active with respect to methanol, ethanol, propanol, and butanol oxidation in H2SO4 solution. The electrochemical measurements also showed that PtSn is more active in ethanol oxidation, whereas PtCu is more active in methanol oxidation. Both materials showed similar electrooxidative activities towards propanol and butanol, and they presented higher electrochemical acidities than pure platinum for alcohol oxidation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alcohol electrooxidation reactions (AERs) have many uses, such as in direct alcohol fuel cells (DAFC), which are electrochemical power generators that show a great deal of promise, especially for portable applications [1]. However, alcohols present slow electrooxidation kinetics, resulting in DAFC power densities that are too low for practical applications [2–4]. Methanol and ethanol are currently two of the most widely studied organic fuels because they are known to produce high power densities in DAFC with platinum-based catalysts. However, during this catalyzed reaction, the platinum surface is poisoned by strongly adsorbed intermediates such as carbon monoxide, due to the dissociative chemisorption of methanol or ethanol [5, 6]. In order to overcome this problem, a second or third metal that is less noble than platinum is added to the catalyst.
The insertion of a less noble metal into the electrocatalyst modifies the electronic structure of platinum and/or produces the oxidized species at a lower potential, which may change the mechanism of alcohol electrooxidation. These alterations are known as the electronic effect [7] and the bifunctional effect [8], respectively, and both speed up the kinetics and decrease the onset potential for alcohol oxidation [9, 10].
In the work reported in the present paper, we studied the use of PtCu, PtSn, and Pt nanoparticles as catalysts in the oxidation of various alcohols (methanol, ethanol, propanol, and butanol) in water solution. PtSn has been studied as a catalyst of alcohol (methanol [11–13] and ethanol [13–16]) oxidation over the last decade; it is currently the most active electrocatalyst used for ethanol oxidation [17, 18] in direct ethanol fuel cells [19], even though the main products of this process are acetaldehyde and acetic acid [20]. In contrast, many different applications have been indicated for PtCu [21, 22]: in the electrocatalysis of DAFC reactions, PtCu has demonstrated activity when used as cathode or an anode. When used as a cathode, Coleman et al. [23] found that PtCu catalyzes the reduction of oxygen in acid and alkaline media. Carbonio et al. [24] found that PtCu could be used as a cathode in direct methanol fuel cells; in that work, PtCu was employed in oxide form following thermal treatment in a reductive atmosphere to increase electrochemical activity and fuel cell performance. PtCu working as anode, Huang et al. [25] reported the use of PtCu with a high density of surface Pt defects for ethanol oxidation, and Magalhães and Colmati [26] found that employing PtCu as a catalyst reduces the onset potential for ethanol oxidation in sulfuric acid solution compared to the application of a pure platinum catalyst; in both cases, PtCu presented high electrochemical activity, indicating that this material can be utilized in a flexible manner in ethanol electrooxidation schemes.
The major difference between PtSn and PtCu is that they show different shifts in the pure Pt lattice structure. When Sn is incorporated into the fcc structure of Pt, the diffraction peaks of the resulting alloy are shifted to slightly lower 2θ values compared to the corresponding peaks for a pure platinum catalyst. In contrast, when Cu is incorporated into the fcc structure of Pt, the diffraction peaks of the resulting alloy are shifted to slightly higher 2θ values compared to the corresponding peaks for a pure platinum catalyst. This indicates that solid solutions of Pt and Sn and of Pt and Cu form, which may lead to different energies of the alcohol molecules adsorbed onto the catalyst and the reaction intermediates as compared to the energies of these molecules on a pure Pt catalyst. Moreover, the introduction of Sn or Cu atoms alters the electron distribution in the orbitals of Pt. Colmati et al. [27, 28] found that a decrease in Pt 5d-band vacancies could be attributed to the donation of electrons from the Sn 5p and s bands to the Pt 5d band, which is a strong acceptor.
Studies of alcohols containing more than two carbons have not explored the electrocatalyzed oxidation of these molecules in acidic media. However, the literature contains reports of studies using Pt single crystals with low and high Müller index values [29] and a supported Pt base electrocatalyst in alkaline media. In addition, Habibi and Dadashpour [30] reported on the oxidation of 2-propanol and 2-butanol on a PtNi alloy in acid media.
Tripković et al. [29] revealed the influence of OH on the platinum surface during the oxidation of alcohols: the OH increases the alcohol electrooxidation current, but Pt–O formation decreases the electrocatalytic activity at very high potential. In addition, OH may poison the surface in some cases. The addition of a less noble metal than platinum, supplying –OH species at lower potential and the OH promote the activity for the alcohol electrooxidation current [31]. Furthermore, solid solution formation alters the electronic structure of the platinum-based electrocatalyst.
Recent studies have focused on isometric alcohols such as 1-propanol and 2-propanol, since these have low onset potentials and only minor poisoning effects [32]. Li et al. [33, 34] studied the mechanism of 1-butanol oxidation by applying FTIR in situ and identified the intermediates involved in electrooxidation. 1-Butanol was found to be the most active of the butanol isomers but exhibited a strong self-poisoning effect along with the formation of CO through butanol dissociation.
The study reported in the present paper focused on the electrooxidation of methanol, ethanol, 1-propanol, and 1-butanol on nanoparticles of PtSn, PtCu, and Pt in acidic media.
Experimental
Electrocatalyst preparation
Carbon-supported PtSn and PtCu electrocatalysts were prepared through the chemical reduction of appropriate precursors in ethanol solution. Carbon powder (Vulcan XC72R, 240 m2 g−1, from Cabot Corporation, Boston, MA, USA) was suspended in a freshwater solution prepared from the metal precursors, i.e., H2PtCl6 (Sigma–Aldrich, St. Louis, MO, USA), SnCl2 (Vetec Química Fina Ltda., Duque de Caxias, Brazil), and CuCl2 (Vetec). This system was then heated to boiling in a reflux system for 3 h. The material was subsequently cooled to room temperature, filtered, washed, and dried out at 60 °C for 2 h.
Energy-dispersive X-ray analysis
The atomic ratios of the PtM/C (M = Cu or Sn) catalysts were established using energy-dispersive X-ray analysis (EDX) coupled to scanning electron microscopy (JSM-6610, JEOL, Tokyo, Japan; NSS Spectral Imaging system, Thermo Scientific, Waltham, MA, USA).
X-ray diffraction
Physical characterization of the materials was carried out using a Shimadzu (Kyoto, Japan) XRD-6000 diffractometer with a scan rate of 0.02° 2θ s−1 between 20° 2θ and 120° 2θ. Part of the catalyst of interest was added to a sample holder which was placed into the diffractometer.
High-resolution transmission electron microscopy
High-resolution transmission electron microscopy (HRTEM) characterization was performed as follows. A carbon film was deposited onto a mica sheet that was placed onto a Cu grid (400 mesh and 3 mm in diameter). The material to be analyzed was dispersed in water through sonication, placed on the carbon film, and left to dry out.
Histograms of particle size were constructed based on measurements of about 300 particles. Measurements were obtained with a technique implemented at the Microscopy Laboratory of the LABMIC (Laboratório de Microscipia Multiusuários da Universidade Federal de Goiás) using a HRTEM microscope (JEM 2100, JEOL) operating at 200 kV and possessing a resolution of 0.2 nm.
Electrochemical measurements
For the electrochemical experiments, the catalysts were ultrasonically dispersed in water with isopropyl alcohol; part of this suspension was stored on top of glass-carbon imbibed in a Teflon rod. We also carried out an experiment without Pt. In this, Vulcan XC72R carbon was ultrasonically dispersed in water with isopropyl alcohol; part of this suspension was stored on top of glass-carbon imbibed in a Teflon rod. The glass-carbon had previously been polished with a diamond paste (1 μm). Finally, the electrode was loaded with 100 μg of Pt and inserted into a glass electrochemical cell with three electrodes: Pt gauze as the auxiliary electrode, Ag/AgCl/Cl −sat as the reference electrode, and the sample as the working electrode. The experiments were carried out in 0.5 mol L−1 sulfuric acid medium as well as at distinct alcohol concentrations. The catalysts were characterized using cyclic voltammetry in the absence and presence of the alcohols of interest, and using chronoamperometry. All of the experiments were carried out at room temperature (27 °C).
Our study also investigated the electrooxidation of methanol, ethanol, 1-propanol, 2-propanol, and 1-butanol on Pt nanoparticles. The prepared working electrode was used for all of the alcohol electrooxidation studies. Before studying a different alcohol, the electrode was washed by immersion in ultrapure water. Cyclic voltammetry performed before each experiment indicated that the electrocatalyst was active.
Results and discussion
The composition of each PtM (M = Cu or Sn) catalyst was assessed via EDX analysis. The EDX-derived compositions of all the catalysts that we investigated are listed in Table 1; the results show that the EDX-derived compositions of the as-prepared PtSn and PtCu catalysts are very close to their nominal compositions.

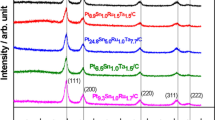
XRD patterns of the carbon-supported PtSn, PtCu, and Pt catalysts prepared by chemical reduction with ethanol reflux
Figure 1 presents the XRD patterns of the as-prepared carbon-supported PtSn, PtCu, and Pt catalysts. The diffraction peak at 20–25° observed in all of the diffraction patterns of the carbon-supported catalysts can be attributed to the (0 0 2) plane of the hexagonal structure of Vulcan XC-72R carbon. As indicated in Fig. 2, all of the XRD patterns clearly show the five main characteristic peaks of the face-centered cubic (fcc) crystalline Pt: the planes (1 1 1), (2 0 0), (2 2 0), (3 1 1), and (2 2 2), demonstrating that all of the catalysts present single-phase disordered structures (i.e., they are solid solutions). These five diffraction peaks are shifted to slightly lower 2θ values for the PtSn catalyst with respect to the corresponding peaks for the Pt/C catalyst [35]. The shifting of the peaks to lower angles indicates the formation of a solid solution of Pt and Sn following the incorporation of Sn into the fcc structure of Pt. In contrast, these five diffraction peaks are shifted to slightly higher 2θ values for the PtCu catalyst with respect to the corresponding peaks for the Pt/C catalyst. The shifting of the peaks to lower angles implies the formation of a solid solution of Pt and Cu following the incorporation of Cu into the fcc structure of Pt [24–26, 36].

HRTEM image at low-magnification (a) and histogram of the particle size distribution (b) for the catalyst PtCu
No peaks from pure Cu or its oxides were observed, but the presence of such peaks cannot be ruled out because pure Cu or its oxides may be present in a very small amount or even in an amorphous form. However, the presence of SnO2 was verified through XRD analysis.
Table 1 presents the lattice parameters of the PtSn, PtCu, and Pt catalysts, which reflect the formation of solid solutions. The lattice parameters obtained for the PtSn catalyst are higher than those obtained for Pt/C, while the lattice parameters obtained for the PtCu catalyst are lower than those found for Pt/C, indicating that the lattice expands after the Pt is alloyed with Sn and contracts after Pt is alloyed with Cu.
The average crystal sizes of the nanoparticles of PtSn, PtCu, and Pt were estimated using Scherrer’s equation d = 0.94 k/Bcosθ [37], where d is the average crystal diameter, k is the wavelength of the X-ray radiation (0.154056 nm), θ is the angle of the (2 2 0) peak, and B is the width in radians of the diffraction peak at half height. The average crystal sizes calculated for all of the catalysts are given in Table 1.
The maximum specific surface areas (S) [38] of the PtSn, PtCu, and Pt nanoparticles were estimated using the equation S = 6 × 10−4/ρd, where S is the surface area (cm2 g−1), and ρ is the density of the metal (Pt = 21.45 g cm−3; Cu = 8.92 g cm−3; Sn = 7.31 g cm−3). d is the average crystal diameter (cm). The surface area (cm2 g−1 PtM) was estimated for each alloy by inserting ρ = Σx i ρ i into the above equation, where x i is the EDX-derived mass content of metal i (i = Pt, Sn/Cu) and ρ i is the density of metal i. The calculated surface areas of the catalysts are given in Table 1.
While the specific area (S) is not the electrochemically active area of the catalyst, it was calculated in this work from the average particle size (i.e., smaller and larger particles can be distinguished in terms of their contributions to the specific area).
When Sn and Cu are added to the Pt lattice, some of the Sn or Cu is in the form of an oxide. The amount of alloyed Sn or Cu (x) and the amount in the oxide form can be calculated with the lattice parameter, as proposed by Antolini at al. [39], assuming that the lattice parameter follows Vergard’s law: x = [(a – a o)/(a s – a o)]x s, where a is the lattice parameter of the synthesized electrocatalyst (values in Table 1), a o is the lattice parameter of Pt/C from E-TEK (a 0 = 0.3911 nm), and a s is the lattice parameter of Pt3M/C from E-TEK (a s = 0.40015 nm, ca. 100 % alloyed, for PtSn; a s = 0.3878 nm, ca. 100 % alloyed, for PtCu). x s is the atomic fraction of Sn in PtSn (0.25) or of Cu in PtCu (0.30). The formation of the solid solution, are given in Table 2. Using the difference between the EDX-derived composition of the catalyst and the value of alloyed non-noble metal (Sn or Cu), we obtained the fractions of unalloyed Sn and Cu. These data are shown in Table 2., these data are not showed in this work.
Figures 2, 3, and 4 show TEM images (subfigure a) and the particle size distributions (subfigure b) of PtCu/C, PtSn/C, and Pt/C.

HRTEM image at low magnification (a) and histogram of the particle size distribution (b) for the catalyst PtSn

HRTEM image at low magnification (a) and histogram of the particle size distribution (b) for the catalyst Pt
As illustrated in Figs. 2a, 3a, and 4a, all of the catalysts presented a homogeneous particle distribution on the carbon support with low particle agglomeration. The particle shape was similar for Pt and PtSn but not for PtCu, which showed irregular particle morphologies such as particles with concave areas and steps. As found by Huang et al. [25], irregular particle morphology increases the electrochemical activity of the nanoparticles. Figures 2b, 3b, and 4b show histograms of the particle size distributions of the catalysts, all of which present a Gaussian shape with the peak at 2.5 nm for PtCu, 3.3 nm for PtSn, and 2.9 nm for Pt. These values were used as the catalyst particle size, as reported in Table 1.
Figure 5 presents cyclic voltammograms for all of the catalysts in sulfuric acid solution as well as the Vulcan XC72-R carbon used as the metal nanoparticle support. As indicated, for all of the electrocatalytic materials, the hydrogen adsorption/desorption region is between −0.2 and +0.1 V vs. Ag/AgCl/Cl −sat , the double-layer region is between 0.1 V and 0.6 V, and the oxide region is above 0.6 V.

Cyclic voltammetry of the materials PtSn, PtCu, and Pt in 0.5 mol L−1 H2SO4. The voltammograms were registered at 20 mV s−1 in ambient temperature
More charge was expected from the adsorption of hydrogen on Pt than on bimetallic materials, but this was not actually observed from cyclic voltammetry (see Fig. 5). This unexpected result can be explained as follows: although the specific surface areas (S) are similar for all of the catalytic materials, their electrochemically active areas are not—the electrochemically active area depends on the exposed surface area, and the frequency of larger particles was greater for Pt than for the alloys, as shown in the histograms in Fig. 4.
The cyclic voltammogram of Vulcan XC72-R carbon presents no peaks in the hydrogen adsorption/desorption and oxide regions (i.e., at low and high potentials), but this curve shows a double-layer region across the full range of the voltammogram; this defines the capacitive current for all of the materials that utilize this carbon support (Pt3Sn, Pt3Cu, and Pt). In the cyclic voltammograms of the electrocatalytic materials, currents that are larger than that of the cyclic voltammogram for carbon can be attributed to the adsorption/desorption of hydrogen and oxygen species. Another important observation is that the carbon is not electrochemically active towards hydrogen and oxygen adsorption/desorption in the potential range studied in acidic media, so there is no contribution from the carbon support to the electrocatalysis of alcohol oxidation. Jow et al. [40] used cyclic voltammetry to determine the surface area of carbon black, and they observed that the influence of the electrolyte (H2SO4) concentration on the double-layer charge was negligible.
Figure 6 provides cyclic voltammograms of all the catalytic materials in sulfuric acid solution with various alcohols (concentration: 1 mol L−1).

Cyclic voltammetry of the materials PtSn, Pt, and PtCu in 0.5 mol L−1 H2SO4 and 0.1 mol L−1 of a methanol, b ethanol, c propanol, and d butanol. The voltammograms were registered at 20 mV s−1 in ambient temperature
The onset potential is the potential at which alcohol oxidation begins. In our study, we considered the onset potential to be the potential when the current is 1 % higher than the current of the cyclic voltammogram obtained without alcohol in the electrolyte. The onset potentials for the catalysts with various alcohols were obtained from Fig. 6 and are shown in Table 3.
To examine the stabilities and electrochemical activities of the catalysts under stationary conditions, chronoamperometry experiments were carried out with different alcohols. These experiments involved polarizing the electrode at 0.3 V vs. Ag/AgCl/Cl −sat and measuring the current from the oxidation of the alcohol of interest. The electrolyte was not stirred and the system was not disturbed. The resulting chronoamperometric data are shown in the “Appendix.”
Figure 7 plots the current observed (at 1800 s) during the oxidation of each alcohol against the number of carbon atoms in the alcohol.

Current (measured at 1800 s) observed during chronoamperometry versus the number of carbon atoms in the alcohol molecule. The open symbols indicate values for isopropanol
We compared the currents recorded at 1800 s for the different alcohols and found that ethanol yielded the highest current when the same electrode was used to oxidize different alcohols. Moreover, it is possible to compare the alcohol oxidation current with increased carbon number in the alcohols molecule of the alcohols and we observe a increase on current from methanol to ethanol but a decreases in the current with increase in the carbon number in the alcohols molecule form ethanol to butanol. Upon comparing the oxidation currents at 1800 s obtained with different catalytic materials for the same alcohol, we observed that PtSn is the most active catalyst for methanol and ethanol oxidation in stationary experiments (see Fig. 7). It is worth comparing these chronoamperometry results with those obtained from cyclic voltammetry at 20 mV s−1 (Fig. 6); according to the cyclic voltammograms, catalysis with PtSn yielded the same current during ethanol and propanol oxidation, while the current decreased with increasing number of carbon atoms in the alcohol when PtCu or Pt was used as the catalyst.
Conclusion
We observed the formation of solid solutions of PtSn and PtCu when these catalysts were prepared using the ethanol reflux method. These nanostructured materials exhibited particle sizes of between 2.5 and 3.5 nm as well as a relatively homogeneous distribution of the carbon support. Electrochemical measurements indicated that PtSn is more electrocatalytically active than PtCu and Pt in ethanol oxidation, whereas PtCu is more active in methanol oxidation. In the electrooxidation of propanol and butanol, these catalysts presented similar electrochemical activities.
References
Kamarudin MZF, Mararudin SK, Masdar MS, Daud (2013) Int J Hydrogen Energy 38:9438–9453
Bonesi A, Asteazaran M, Moreno MS, Zampieri G, Bengio S, Triaca W, Castro Luna AM (2013) J Solid State Electrochem 17:1823–1829
Linares JJ, Zignani SC, Rocha TA, Gonzalez ER (2013) J Appl Electrochem 43:147–158
Pearson A, O’Mullane AP (2015) Chem Commun 51:11297–11300
Shao MH, Adzic RR (2005) Electrochim Acta 50:2415–2422
Silva JCM, Anea B, de Souza RFB, Assumpção MHMT, Calegaro ML, Neto AO, Santos MC (2013) J Braz Chem Soc 24:1553–1560
Coutanceau C, Brimaud S, Lamy C, Leger J-M, Dubai L, Rousseau S, Vigier F (2008) Electrochim Acta 53:6865–6880
Ciapina E, Gonzalez ER (2009) J Electroanal Chem 626:130–142
Asgardi J, Calderon JC, Alcaide F, Quereja A, Calvillo L, Lazaro MJ, Garcia G, Pastor E (2015) Appl Catal B Environ 168:33–41
de Souza RFB, Silva JCM, Assumpção MHT, Neto AO, Santos MC (2014) Electrochim Acta 117:292–298
Colmati F, Antolini E, Gonzalez ER (2005) Electrochim Acta 50:5496–5503
Liu Z, Guo B, Hong L, Lim TH (2006) Electrochem Commun 8:83–90
Aricò AS, Creti P, Antonucci PL, Antonucci V (1998) J Electrochem Soc 2:66–68
Lamy C, Belgsir EM, Léger JM (2001) J Appl Electrochem 31:799–809
Zhou WJ, Song SQ, Li WZ, Zhou ZH, Sun GQ, Xin Q, Douvartzides S, Tsiakaras P (2005) J Power Souces 140:50–58
Kowal A, Gojkovic SL, Lee K-S, Olszewski P, Sung Y-E (2009) Electrochem Commun 11:724–727
Meenakshi S, Sridhar P, Pitchumani S (2014) RCS Adv 4:44386–44393
Queiroz AC, Silva WO, Rodrigues IA, Lima FHB (2014) Appl Catal B Environ 160–161:423–435
Na L, Zhao TS, Li YS (2015) Renew Sust Energ Rev 50:1462–1468
Beyhan S, Coutanceau C, Leger J-M, Napporn TW, Kardirgan F (2013) Int J Hydrogen Energy 38:6830–6841
Lee M, Hwang Y, Yun k-H, Chung Y-C (2015) J Power Sources 288:296–301
Liu J, Zhao O, Chem S, Yi L, Wang X, Wei W (2015) Electrochim Acta 171:96–104
Coleman EJ, Chowdhury MH, Co AC (2015) ACS Catal 5:1245–1253
Carbonio EA, Colmati F, Ciapina EG, Pereira ME, Gonzalez ER (2010) J Braz Chem Soc 21:590–602
Huang M, Jiang Y, Jin C, Ren J, Zhou Z, Guan L (2014) Electrochim Acta 125:29–37
Magalhães MM, Colmati F (2014) J Braz Chem Soc 25:1317–1325
Colmati F, Antolini E, Gonzalez ER (2007) J Electrochem Soc 154:B39–B47
Román-Martinez MC, Cazorla-Amorós D, Miguel S, Scelza O (2004) J Jpn Pet Inst 47:164–178
Tripković AV, Popović KDJ, Lović JD (2001) Electrochim Acta 46:3163–3173
Habibi B, Dadashpour (2013) Electrochim Acta 88:157–164
Watanabe M, Motoo S (1975) J Electroanal Chem 60:275–279
Lee C-G, Umeda M, Uchida I (2006) J Power Sources 160:78–89
Li N-H, Sun S-G (1998) J Electroanal Chem 448:5–15
Li N-H, Sun S-G (1997) J Electroanal Chem 436:65–72
Colmati F, Antolini E, Gonzalez ER (2007) Appl Catal B Environ 73:106–115
Liu L, Samjeské G, Takao S, Nagasawa K, Iwasawa Y (2014) J Power Souces 253:1–8
Ascarelli P, Contini V, Giorgi R (2002) J Appl Phys 91:4556–4561
Starz KA, Auer E, Lehmann T, Zuber R (1999) J Power Sources 84:167–172
Antolini E, Colmati F, Gonzalez ER (2009) J Power Sources 193:555–561
Jow J-J, Hsieh L-Y, Cho H-P, Chen H-R, Kuo C-W (2013) J Ind Eng Chem 19:1730–1734
Acknowledgments
The authors would like to thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Proc. 554569/2010-8 and Proc. 475609/2008-5) for financial support as well as the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) for granting our scholarship. We would also like to thank Tatiane Oliveira dos Santos from the Microscopy Laboratory (LABMIC) of the Federal University of Goiás, Goiânia, Brazil, for permitting the use of the microscope (a JEM-2010 HRTEM microscope, JEOL, Tokyo, Japan).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors state that there is no conflict of interest associated with this work.
Appendix
Appendix
As illustrated in Fig. 8, the alcohol oxidation current was relatively stable at 1800 s, except for isopropanol. This behavior can be attributed to differences in the adsorption of the alcohols on the electrode surface. Some studies have found that small and linear alcohols such as methanol and ethanol are adsorbed at a platinum electrode in an acidic medium via the α-carbon in the alcohol molecule. Propanol and butanol are also adsorbed in this manner, but the α-carbon is shielded by methyl groups in isopropanol, so only weak adsorption occurs on the Pt surface and the oxidation current does not stabilize within 1800 s. At this section, the scale of the y-axis of the graphics was not similar for all figures to emphasize the behavior of the curves with time once the current values are very different.

Chronoamperometric curves (obtained at 0.3 V vs Ag / AgCl/Cl− sat) for a methanol, b ethanol, c propanol, and d butanol on Pt3Sn/C, Pt3Cu/C, and Pt/C electrodes in 0.5 mol L−1H2SO4
Rights and permissions
About this article

Cite this article
dos Reis, R.G.C.S., Colmati, F. Electrochemical alcohol oxidation: a comparative study of the behavior of methanol, ethanol, propanol, and butanol on carbon-supported PtSn, PtCu, and Pt nanoparticles. J Solid State Electrochem 20, 2559–2567 (2016). https://doi.org/10.1007/s10008-016-3323-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-016-3323-3