
Abstract
We have studied LiFePO4/C nanocomposites prepared by sol-gel method using lauric acid as a surfactant and calcined at different temperatures between 600 and 900 °C. In addition to the major LiFePO4 phase, all the samples show a varying amount of in situ Fe2P impurity phase characterized by x-ray diffraction, magnetic measurements, and Mössbauer spectroscopy. The amount of Fe2P impurity phase increases with increasing calcination temperature. Of all the samples studied, the LiFePO4/C sample calcined at 700 °C which contains ∼15 wt% Fe2P shows the least charge transfer resistance and a better electrochemical performance with a discharge capacity of 136 mA h g−1 at a rate of 1 C, 121 mA h g−1 at 10 C (∼70 % of the theoretical capacity of LiFePO4), and excellent cycleability. Although further increase in the amount of Fe2P reduces the overall capacity, frequency-dependent Warburg impedance analyses show that all samples calcined at temperatures ≥700 °C have an order of magnitude higher Li+ diffusion coefficient (∼1.3 × 10−13 cm2 s−1) compared to the one calcined at 600 °C, as well as the values reported in literature. This work suggests that controlling the reduction environment and the temperature during the synthesis process can be used to optimize the amount of conducting Fe2P for obtaining the best capacity for the high power batteries.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Lithium iron phosphate (LiFePO4) has been used as a cathode material in commercially available rechargeable batteries for nearly two decades. LiFePO4 is intensely investigated since it was proposed by Padhi et al. [1] as a possible cathode material for Li-ion rechargeable batteries. It is one of the most promising cathode material for the next generation Li-ion batteries for military, electric vehicles, and aerospace applications, due to its high theoretical capacity (170 mA h g−1), availability of inexpensive starting materials, its environmental friendliness, and excellent thermal stability [2–6]. However, the main limitation of LiFePO4 has been its low electronic conductivity (∼10−9 S cm−1), which leads to high impedance, low capacity, and low rate capability. Many researchers have been investigating on improving the performance and safety aspects of LiFePO4. The methods of improving the performance including carbon coating [7–10], particle size reduction [11–13], and metal doping [14–16] have been proposed. Incorporation of conducting metal fluoride phases such as FeF2 into LiFePO4 is reported to be a viable method for enhancing the electronic conductivity of this material [14, 17, 18]. The presence of iron phosphide (Fe2P) has also been found to increase the performance of Li-ion batteries [15].
Fe2P is known to be a very good electronic conductor [15]. An olivine phosphate, particularly LiFePO4, has been known to undergo carbothermal reduction at high temperatures to form Fe2P and other metal phosphides. In presence of a suitable carbon-containing surfactant, at high temperatures, carbon reacts with LiFePO4 to form CO2 or CO, which leads to the formation of a two-phase system: Li3PO4/Fe2P [19]. Thus, the conducting Fe2P phase can form intrinsically in LiFePO4. Fe2P-incorporated LiFePO4 has shown an improvement in electrochemical performance, as it provides a conducting network that enhances the electron transport through the material. It has been known that even a small amount of Fe2P can enhance the electronic conductivity by 4 orders of magnitude [20]. However, excessive amount of this conducting phase may result in loss of capacity, as it forms at the expense of LiFePO4, which is responsible for lithium insertion/de-insertion reactions [21]. There are several studies which have reported the effect of addition of Fe2P on the electrical and electrochemical properties of LiFePO4 [22–24]. Liu et al. [25] prepared LiFePO4 by co-precipitation method using polyacrylamide as carbon source that provided a reducing environment for the formation of Fe2P. They found Fe2P impurity was formed at 750 °C, and LiFePO4 with Fe2P impurity exhibited better electronic conductivity and improved electrochemical performance. Although the amount of Fe2P was not reported, they found the amount of Fe2P increased with increasing calcination time. Rahman et al. [26] synthesized LiFePO4-Fe2P-C material using solvent-assisted solid-state reaction with various amounts of citric acid as a source of carbon. They found that the use of solvent assists in the formation of Fe2P in the proximity of LiFePO4 particles, which provides a percolating network resulting in a very high electronic conductivity. They claimed a very high capacity 136 mA h g−1 at 10 C rate. Kim et al. [27] prepared LiFePO4/Fe2P composites by mechanical alloying in order to improve the electrical conductivity. The sample containing 8 % of Fe2P in LiFePO4/Fe2P composite showed a high capacity ∼160 mA h g−1 at rate of C/20, and ∼110 mA h g−1 at a rate of 1 C with good cycleability. Rho et al. [28] studied the surface chemistry of four LiFePO4 samples, prepared by heat treatment under increasing reducing environment, using Mössbauer and x-ray photoelectron spectroscopy. When the samples were treated for a longer period from 15 to 24 h in 7 % H2/N2 atmosphere at 600 °C, the authors found Fe2P impurity phase between 4 and 18 wt%, along with Li3PO4 formed on the surface of the parent LiFePO4. Their results suggest that an optimum amount, between 5 and 10 wt%, of Fe2P gives rise to a greatly enhanced electrochemical performance of the composite. Another study reported that the presence of FeP is more favorable over Fe2P, and indicated that 16 % of Fe2P greatly lowered the discharge capacity [29].
From the review of the literature, it is clear that the presence of Fe2P phase influences the performance of LiFePO4 cathode. However, several questions still remain unanswered. It is important to know (1) how does the calcination temperature affect the amount of Fe2P phase produced in a reducing environment? (2) what are the effects of the presence of Fe2P impurity phase on diffusion of Li+ ions through LiFePO4 and the electrochemical performance of LiFePO4 cathode material? and (3) what is the optimum amount of Fe2P required to obtain the best performance of LiFePO4 cathode? To answer these questions, we have performed an extensive temperature-dependent study on LiFePO4/C prepared by a simple and cost-effective sol-gel method followed by calcination in reducing Ar/H2. We have synthesized samples at various temperatures between 600 and 900 °C and quantified the Fe2P content (5–37 wt%) using magnetic and Mössbauer spectroscopy measurements. We found that the sample calcined at 700 °C yields an optimum amount of Fe2P (15 wt%) phase that shows an enhanced capacity with high rate capability of 136 mA h g−1 at rate of 1 C and 121 mA h g−1 at 10 C. We further demonstrate that although the excessive amount of Fe2P decreased the overall discharge capacity, all samples calcined at temperatures ≥700 °C have an order of magnitude higher Li+ diffusion coefficient (∼1.3 × 10−13 cm2 s−1) compared to the one calcined at 600 °C as well as the values reported in literature.
Experimental details
Synthesis
LiFePO4/C samples were synthesized by the sol-gel method [29]. CH3COOLi·2H2O, 99 % (Alfa Aesar), FeCl3 (Fisher Scientific), and P2O5 (Fisher Scientific) were used as precursors. FeCl3 and P2O5 were dissolved in 200 proof ethanol in two separate beakers, then mixed and stirred for 3 h in a three-neck flask under constant flow of nitrogen. Then lithium acetate dissolved in ethanol was added to the solution and was allowed to stir for additional 3 h. Lauric acid dissolved in ethanol was added as a source of carbon and the solution was kept for 3 more hours of stirring under the same conditions and the resulting solution was dried at 90 °C to form a dry gel, which was subsequently calcined at 600, 700, 800, and 900 °C for 10 h under the flow of Ar/H2 (90 %/10 %). Carbon content of the samples was measured by CHN elemental analyses, where the sample is combusted in a pure oxygen environment and found to be ∼4 % in the first three samples and ∼2 % in the fourth sample. Hereafter, these samples will be referred as LFP-600, LFP-700, LFP-800, and LFP-900, respectively.
Characterization
X-ray diffraction (XRD) measurements were performed using a Rigaku Minflex-600 diffractometer using Cu Kα (λ = 1.54 Å) x-rays to determine the phase purity of the samples. Electrical conductivity of the composite samples was measured by preparing pressed pellets and attaching electrodes using conducting silver epoxy in a Van der Pauw geometry. Magnetic measurements were done using a Physical Properties Measurement System (PPMS). Temperature-dependent ac magnetization measurements were done at 500 Oe and saturation magnetization (M) was measured by varying the magnetic field (H) from −5 to +5 T. The morphology of the samples was investigated using JSM-6510-LV-LGS SEM and JEOL 2010 TEM.
57Fe Mössbauer spectroscopy
57Fe Mössbauer spectra were recorded in the transmission geometry using both sides of a (Wissel) transducer coupled to 57Co in Rh matrix source of about 50 mCi and 256 channels of a multichannel analyzer. The velocity calibration and the linearity verification were performed using a thin iron foil. For Mössbauer measurements, approximately 70 mg of the sample was uniformly distributed in a Teflon circular cell of 1.7 cm diameter. The isomer shift values are reported with reference to α-Fe foil. The spectra were least square fitted with MossWin program.
Electrochemical measurements
The electrochemical characterization of the samples was performed in a standard coin cell geometry with lithium metal as an active anode. The active cathode materials and Super P as a conducting material were mixed (95:5 ratio) and ground for 20 min (without a binder). The homogenous mixture was put on an aluminum mesh (current collector) and was then hand pressed between two steel cylinders [30]. The prepared cathode was cycled against Li metal electrode as a counter electrode separated by a polymeric separator soaked in a binary electrolyte consisting of ethylene carbonate (EC) and dimethyl carbonate (DMC) (50:50) containing 1 M LiPF6. The room temperature galvanostatic charge and discharge measurements were carried out at different C-rates in the voltage range 2.2–4.2 V. Electrochemical impedance spectroscopy measurements were carried out using a Gamry electrochemical system in the frequency region of 0.01 Hz–100 kHz with AC amplitude 10 mV.
Results and discussion
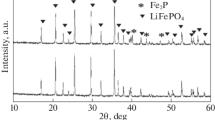
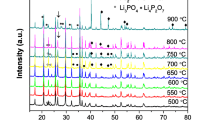
Figure 1 shows the XRD patterns of all LiFePO4 samples calcined at 600–900 °C for 10 h. All the observed Bragg peaks in the LFP-600 sample can be indexed to the olivine LiFPO4 structure with a space group Pnma, which implies the absence of any detectable impurity phase. Additional peaks, which were indexed to iron phosphide (Fe2P) and Li3PO4, are observed in the XRD patterns of samples annealed at 700 °C and higher, and the amount of these phases steadily increases with calcination temperature. These impurity phases result from carbothermal reduction of LiFePO4 at higher temperatures in reducing environment provided by the lauric acid (carbon source) and the flowing forming gas. As the formation of Li3PO4 requires three Li ions, this phase does not grow as quickly as Fe2P because dissociation of LiFePO4 is not fast enough up to 700 °C. Only at calcination temperatures of >700 °C their amount begins to appear considerably according to following carbothermal and/or hydrogen reduction reactions [28]:

XRD patterns of LiFePO4 samples annealed for 10 h
In order to find the weight fraction of impurity phases, we have performed Rietveld refinement of XRD data of LFP-600, LFP-700, LFP-800, and LFP-900 samples. As an example, we show the Rietveld fitting of XRD data of LFP-700 sample in Fig. 2. The crystallite size of LiFePO4, obtained from Rietveld fitting, showed an increase from ∼99 nm in LFP-600 to ∼125 nm in LFP-900, as expected. Further, Table 1 lists the weight percentage of LiFePO4, Li3PO4, and Fe2P in the samples studied in this work. We note that amount of Fe2P increases with the calcination temperature, but the weight ratio between Fe2P and Li3PO4 is less than the expected ratio of ∼2:1 (molar ratio 3:2) in first three samples, very similar to the observations in ref. [28], implying the presence of amorphous or nano-sized impurity phases. The room temperature electrical conductivity measured on the pressed pellets of the LFP-700, LFP-800, and LFP-900 samples showed 2 orders of magnitude higher conductivity (0.8–2.0 × 10−1 S cm−1) compared to that of LFP-600 sample (∼2 × 10−3 S cm−1) which is attributed to the presence of conducting network of Fe2P impurity phase.

Rietveld fitting of XRD pattern of LFP-700 sample
Figure 3 shows SEM images of the samples. The grain size of LFP-600 and LFP-700 samples appears to be very similar. Both of them contain grains of uniform size, estimated to be ∼100 nm, with some agglomeration. However, the samples calcined at higher temperatures (LFP-800 and LFP-900) show noticeably larger grain size and increased nonuniform size distribution (Fig. 3c, d). The EDXS measurements on the LFP-900 sample showed relatively more Fe-rich regions (see inset Fig. 3d), indicating the presence of more Fe2P regions compared to LFP-600 sample (see inset Fig. 3a).

SEM images of LiFePO4 samples: a LFP-600; b LFP-700; c LFP-800; and d LFP-900. The scale bar is 1 μm. The insets in a and b show EDX spectra as described in the text
We further investigated the particle morphology and size distribution using TEM. Figure 4 shows the TEM images of LFP-700 sample at two different magnifications, revealing particles in the size range of ∼80–100 nm. The particles show a rough morphology due to decomposition of LiFePO4 into Fe2P and Li3PO4 on the surface. The arrow marks in Fig. 4a show the inter-particle regions consisting of possibly carbon, Fe2P, and Li3PO4 very similar to the observations reported in ref. [26]. Under higher magnification, the particle surface further reveals the formation of sub-nanoregions of 2–4 nm (see Fig. 4b) of the decomposed products, which may not be detected by XRD but could influence the electrochemical properties of the composite material [26].

TEM images of LFP-700 sample at two different magnifications. The arrow marks in a show inter-particle regions consisting of possibly carbon, Fe2P, and Li3PO4; b magnified image of the circled region showing the sub-nano features on the surface of the particle
Since Fe2P is magnetic with a first order ferromagnetic transition at ∼220 °C [31, 32], we measured the magnetic properties to confirm its presence in the samples. Figure 5a shows the zero field cooled (ZFC) temperature dependence of magnetization at an applied field of 500 Oe and Fig. 5b shows the measured hysteresis loops of magnetization vs applied field for the samples. Although Fe2P is not detectable in the XRD (Table 1), its first order ferromagnetic transition at ∼220 °C is seen in all the samples, albeit it is rather weak in LFP-600, whereas it is quite clear in samples calcined at higher temperatures due to the presence of considerable amount of Fe2P. Thus, the magnetization measurements confirm the presence of Fe2P in all the samples and demonstrate an increase in the amount of Fe2P with increasing calcination temperature (see Fig. 5b). Although the magnetization is not saturated at an applied field of ±10 kOe, the maximum magnetization achieved in each sample was compared with the saturation magnetization value, 32.1 emu g−1, observed for nanocrystallites of Fe2P reported in literature [33]. Considering this value as 100 %, the estimated amount of Fe2P in LFP-600, LFP-700, LFP-800, and LFP-900 samples is 6, 12, 28, and 56 %, respectively. As a result, the amount of active material for Li insertion/de-insertion is substantially reduced in LFP-800 and LFP-900, which is expected to lower the overall specific capacity.

a Magnetization vs. temperature of LiFePO4 samples; b hysteresis loops of LiFePO4 samples measured at 10 K
To further confirm the amount of Fe2P, as determined by magnetization measurements, Mössbauer spectra (see Fig. 6) of the samples were recorded. Summary of the Mössbauer parameters is given in Table 2. The 57Fe Mössbauer spectra of LiFePO4 with Fe2P impurity consists of three quadrupole doublets. First, a symmetric and dominant doublet with an isomer shift ∼1.22 mm/s and a quadrupole splitting of ∼2.97 mm/s arises due to high spin configuration of 3d electrons in Fe2+ ion and charge asymmetry around Fe in parent LiFePO4 [28, 34]. The second doublet with an isomer shift of 0.61 mm/s and a quadrupole splitting of 0.43 mm/s is due to Fe3+ occupying 3f site in Fe2P and the third doublet with an isomer shift of 0.19 mm/s and a quadrupole splitting of 0.1 mm/s is due to Fe3+ at the pyramidal 3 g site for Fe2P structure [28, 35]. In the present study, we did not observe any FeP phase as reported in ref. [28]. It is reasonable to assume that the FeP phase may have transformed into Fe2P phase due to higher annealing temperatures and reducing environment. A very small signature of one of the two doublets for Fe2P in LFP-600 indicates an insignificant amount of Fe2P occupying Fe-3f site only. We have estimated the amount of Fe2+ and Fe3+ from the relative area under the respective peaks in the Mössbauer spectra. The percentage of Fe2+ and Fe3+ phases in the samples is given in Table 2, and corresponding mol% and wt% of LiFePO4, Fe2P, and Li3PO4, calculated using Eq. 1, are listed in Table 3. It is interesting that the total amount of Fe2P calculated from the Mössbauer spectroscopy (Table 3) and magnetic measurements are close to each other in first three samples, but differ from those estimated by Rietveld refinement of XRD data (Table 1). As explained in a previous section, this is due to the presence of amorphous or sub-nanoregions of Fe2P and Li3PO4 as shown in TEM images (Fig. 4).

Mössbauer spectra of LiFePO4 samples measured at room temperature
The galvanostatic charge–discharge voltage profiles of the samples measured at a rate of 1 C are shown in Fig. 7. The observed capacity of 136, 102, and 78 mA h g−1, for LFP-700, LFP-800, and LFP-900 samples, respectively, are very close to the expected capacity calculated using the wt% of LiFeO4 (Table 2) deduced from Mössbauer measurements. However, LFP-600 sample shows a significantly lower capacity of 120 mA h g−1 compared to the expected capacity of 158 mA h g−1. All the samples show a potential plateau at ∼3.4 V corresponding to Fe2+/Fe3+ redox couple. The potential plateau of LFP-700 is wider compared to the other samples, which indicates that Li can be inserted and de-inserted more efficiently.

Charge–discharge profiles of LiFePO4 samples measured at a rate of 1 C
Figure 8a shows the impedance spectra (Nyquist plots) of the samples. The spectra display typical characteristics of an electrochemical cell consisting of a small intercept in the higher frequency region, a depressed semicircle in the intermediate frequency region, and an inclined line in the low-frequency region. The small intercept corresponds to the ohmic resistance, representing the resistance of the electrolyte. The depressed semicircle is related to the charge transfer resistance and the double layer capacitance between the electrolyte and cathode. The inclined line is related to the frequency-dependent Warburg impedance associated with Li+ ion diffusion in the cathode active particles. The values for the charge transfer resistance (R ct) extracted from fitting the impedance data as described below are listed in Table 4. LFP-700 exhibits the lowest charge transfer resistance and hence highest specific capacity suggesting that the amount of Fe2P present in this sample may be the optimum amount to provide highly conducting network, which enhances the electron transport in the sample. Although the samples LFP-800 and LFP-900, calcined at 800 and 900 °C, have lower charge transfer resistance compared to the LFP-600 sample, their specific capacity is lower due to the presence of less active material for Li intercalation and deintercalation process.

a Nyquist plots of LiFePO4 samples; b a plot of the Z′ vs. ω −1/2 in the low-frequency region
To further understand the above behavior, the diffusion coefficient of lithium ion (D Li) was determined by the Z′ dependence on ω in the low-frequency region, which is described by [36],
where, σ is the Warburg coefficient and R s and R ct are the solution and the charge transfer resistances. σ is related to D Li by,
where, R is the gas constant, T is the absolute temperature, A is the surface area of the cathode (0.28 cm2 in our case), n is the number of electrons per molecule during oxidation, F is the Faraday constant, and C Li is the concentration of lithium ion (0.0228 mol/cm3). As expected, a plot of Z′ versus ω −1/2 (see Fig. 7b) shows linear behavior with values of slope σ. The diffusion coefficient and apparent exchange current density (I o) [36],
along with other relevant parameters for the samples are given in Table 4. The LFP-600 sample has the lowest diffusion coefficient (3.5 × 10−14 cm2 s−1) while LFP-700, LFP-800, and LFP-900 samples have similar value, (1.3 ± 0.1) × 10−13 cm2 s−1, which is about an order of magnitude higher than that of LFP-600 and values reported in the literature [37–39]. Of four samples studied in this work, the LFP-700 sample has the least R ct and the highest exchange current density compared to other carbon-coated LiFePO4 samples reported in literature [40], perhaps due to the presence of conducting nanoregions of Fe2P on the surface LiFePO4 particles. This result corroborates with an earlier study on LiFePO4/Fe2P-C composite cathode [26].
Figure 9 shows the discharge capacity y of the LiFePO4 samples at various rates from 1 to 10 C. Clearly, the LFP-700 sample not only shows the best capacity at 1 C rate but also retains more than 70 % of its capacity when tested at higher rate of 10 C (faster charging and discharging). This sample also retains nearly 100 % of its initial capacity even after 350 cycles at a rate of 5 C (see inset in Fig. 9). We attribute the enhanced electrochemical performance of LFP-700 to faster kinetics of the cell reactions (higher diffusion constant) along with higher catalytic activity (higher exchange current density) compared to other samples. We have also calculated the characteristic diffusion length, \( {L}_{\max, \mathrm{L}\mathrm{i}}=\sqrt{\tau\;D{}_{\mathrm{Li}}} \) [41], where τ is the diffusion time constant. It has been shown that at higher C-rate (faster charging/discharging), the characteristic diffusion length decreases with diffusion time constant (taken as the discharging time) [38], and the particle size comparable to the characteristic diffusion length is indicative of better electrochemical performance at higher charge/discharge rates. The estimated characteristic diffusion length for LiFePO4 samples annealed at higher temperatures, using diffusion time constant equivalent to the discharging time for 10 C rate, to be ≈ 70 nm. This means we can obtain maximum possible capacity from the sample, after accounting for inactive components, at lower charging/discharging rate of 1 C. This result is in agreement with our observations (see Table 3).

Capacity of LiFePO4 samples during continuous cycling at different charging rates. The inset shows the capacity retention for LFP-700 sample at 5 C rate
Conclusions
In summary, we have prepared several LiFePO4/C samples calcined at different temperatures between 600 and 900 °C and studied their structural, magnetic, and electrochemical properties. The impurity phase Fe2P was analyzed by XRD, magnetic measurements, and Mössbauer spectroscopy. We found that the amount of the impurity phase strongly depends on the calcination temperature. LiFePO4/C samples calcined at 600 and 700 °C consist of LiFePO4 particles with size ∼90–100 nm and contain 5 % and 16 wt% Fe2P phase. The amount of Fe2P grows considerably in the samples calcined at 800 and 900 °C. Comparing all the properties of the LiFePO4/C samples, the sample calcined at 700 °C exhibits better electrochemical performance with a capacity of 136 mA h g−1 at 1 C, 121 mA h g−1 at 10 C (∼70 % of theoretical capacity of LiFePO4), and excellent cycleability. With ∼15 wt% of Fe2P, it exhibits the least charge transfer resistance of 43 Ω with an electronic conductivity of ∼10−1 s cm−1 and Li+ diffusion coefficient of ∼1.3 × 10−13 cm2 s−1. This indicates the importance of controlling both the reduction environment and temperature during the synthesis process to produce the proper amount of Fe2P phase necessary for enhancing the electrochemical performance of LiFePO4/C nanocomposites.
References
Padhi AK, Nanjundaswamy K, Goodenough JB (1997) J Electrochem Soc 144:1188–1194
Ellis BL, Lee KT, Nazar LF (2010) Chem Mater 22:691–714
Scrosati B, Garche J (2010) J Power Sources 195:2419–2430
Huang H, Faulkner T, Barker J, Saidi MY (2009) J Power Sources 189:748–751
Tarascon JM, Recham N, Armand M, Chotard JN, Barpanda P, Walker W, Dupont L (2010) Chem Mater 22:724–739
Wang YG, He P, Zhou HS (2011) Energy Environ Sci 4:805–817
Julien CM, Zaghib K, Mauger A, Groult H (2012) Adv Chem Eng Sci 2:321–329
Doeff MM, Wilcox JD, Kostecki R, Lau G (2006) J Power Sources 163:180–184
Dominko R, Bele M, Gaberscek M, Remskar M, Hanzel D, Pejovnik S, Jamnik J (2005) J Electrochem Soc 152:A607–A610
Dominko R, Bele M, Goupil JM, Gaberscek M, Hanzel D, Arcon I, Jamnik J (2007) Chem Mater 19:2960–2969
Delmas C, Maccario M, Croguennec L, Le Cras F, Weill F (2008) Nat Mater 7:665–671
Gibot RP, Casas-Cabanas M, Laffont L, Levasseur S, Carlach P, Hamelet S, Tarascon JM, Masquelier C (2008) Nat Mater 7:741–747
Hsu KF, Tsay SY, Hwang BJ (2004) J Mater Chem 14:2690–2695
Croce F, D’Epifanio A, Hassoun J, Deptula A, Olczac T, Scrosati B (2002) Electrochem Solid State Lett 5:A47–A50
Herle PS, Ellis B, Coombs N, Nazar LF (2004) Nat Mater 3:147–152
Meethong N, Kao YH, Speakman SA, Chiang YM (2009) Adv Funct Mater 19:1060–1070
Huang H, Yin SC, Nazar LF (2001) Electrochem Solid -State Lett 4:A170–A172
Chung SY, Bloking JT, Chiang YM (2002) Nat Mater 1:123–128
Ellis B, Herle PS, Rho YH, Nazar LF, Dunlap R, Perry LK, Ryan DH (2007) Farad Discuss 134:119–141
Lee KT, Lee KS (2009) J Power Sources 189:435–439
Song MS, Kim DY, Kang YM, Kim YI, Lee JY, Kwon HS (2008) J Power Sources 180:546–552
Xu Y, Lu Y, Yan L, Yang Z, Yang R (2006) J Power Sources 160:570–576
Qiu Y, Geng Y, Yu J, Zuo X (2014) J Mater Sci 49:504–509
Liu H, Xie J, Wang K (2008) Solid State Ionics 179:1768–1771
Liu Y, Cao C, Li J, Xu X (2010) J Appl Electrochem 40:419–425
Rahman MM, Wang J, Zeng R, Wexler D, Liu HK (2012) J Power Sources 206:259–266
Kim CW, Park JS, Lee KS (2006) J Power Sources 163:144–150
Rho YH, Nazar LF, Perry L, Ryan D (2007) J Electrochem Soc 154:A283–A289
Lin Y, Gao MX, Zhu D, Liu YF, Pan HG (2008) J Power Sources 184:444–448
Dhindsa KS, Mandal BP, Bazzi K, Lin MW, Nazri M, Nazri GA, Naik VM, Garg VK, Oliveira AC, Vaishnava P, Naik R, Zhou ZX (2013) Solid State Ionics 253:94–100
Wappling R, Haggstrom L, Ericsson T, Devanarayanan S, Karlsson E, Carlsson B, Rundqvist S (1974) J De Physique 35:C6–597
Muthuswamy E, Kharel PR, Lawes G, Brock SL (2009) ACS Nano 3:2383–2393
Luo F, Su HL, Song W, Wang ZM, Yan ZG, Yan CH (2004) J Mater Chem 14:111–115
Yamada A, Chung SC, Hinokuma K (2001) J Electrochem Soc 148:A224–A229
Ericsson T, Haggstrom L, Wappling R, Methasiri (1980) Physical Scripta 21:212–216
Bard AJ, Faulker LR (2001) Electrochemical Methods-Fundamental and Applications, 2ndEd. Wiley, New York
Prosini PP, Lisi M, Zane D, Pasquali M (2002) Solid State Ionics 148:45–51
Kumar A, Thomas R, Karan NK, Saavedra-Arias JJ, Singh MK, Majumder SB, Tomar MS, Katiyar RS (2009) J Nanotech 2009: Article ID 176517, Doi:10.1155/2009/176517
Yu D, Fietzek C, Weydanz W, Donoue K, Inoue T, Kurokawa H, Fujitani S (2007) Electrochem Soc 154:A253–A257
Pang L, Zhao M, Zhao X, Chai Y (2012) J Power Sources 201:253–258
Levi MD, Lu Z, Aurbach D (2001) Solid State Ionics 143:309–318
Acknowledgments
We thank the Richard Barber Foundation for financial support to perform this work. We also thank Dr. Federico Rabuffetti at Wayne State University for useful discussions regarding Rietveld fitting.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Dhindsa, K.S., Kumar, A., Nazri, G.A. et al. Enhanced electrochemical performance of LiFePO4/C nanocomposites due to in situ formation of Fe2P impurities. J Solid State Electrochem 20, 2275–2282 (2016). https://doi.org/10.1007/s10008-016-3239-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-016-3239-y