
Abstract
Nanocrystals (NCs) of a Pt or Pd or Pt–Pd alloy shell supported on a Au core were synthesized and dripped onto a glassy carbon (GC) surface to generate thin films. Their electrocatalytic activity towards the oxygen reduction reaction (ORR) was studied employing hydrodynamic cyclic voltammetry. Of the Pt or Pt–Pd alloy electrocatalysts synthesized over a Au core (including Au cores produced from redox-transmetalation of Ni cores), Pt–Pd@Au, Pt@Au, and Pt@Au′ NCs—which contained heterogeneous NCs with spherical, triangular, squared, pentagonal, hexagonal, heptagonal, and rod-like shapes, with large (about 80 nm in several cases), well-defined crystalline structures, and evidenced a nanodendritic Pt or Pt–Pd alloy covering pattern at the NC surface—exhibited high electrocatalytic activity towards ORR and high stability (without dissolution of inner metallic nanoparticles such as Au) after 10,000 potential scans—features that suggest their utility for use in acid fuel cells.

Pt–Pd@Au, Pt@Au, and Pt@Au′ NCs exhibited high electrocatalytic activity towards ORR and high stability (without dissolution of inner metallic nanoparticles such as Au) after 10,000 potential scans.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As regards the oxygen reduction reaction (ORR)—crucial in energy-related electrochemical applications such as fuel cells—Au catalysis is inferior to highly active metals such as Pt—the most reactive metal in this context [1]. In addition, fast degradation of carbon-based systems, owing to a lack of chemical interaction between Pt particles and carbon support, as well as the occurrence of corrosion and fast contamination of the carbon support, asks for further research on alternative strategies [1, 2]. Combining Pt with other metal elements to form bi- or multimetallic nanostructures could prove efficient in tuning the structure and catalytic activity of Pt [3].
Bimetallic systems provide further means of tuning the properties of nanoparticles by varying composition and chemical ordering within nanoparticles [4]. In particular, Au–Pt [3–5] and Au–Pd nanoparticle systems have attracted interest because of their potential applications as catalysts. Moreover, their composition and the distribution of each metal have been shown to impact nanoparticle activity and selectivity [4].
In recent years, the skyrocketing prices of Pt (which more than tripled over the past few decades and reached ca. US$1600/oz in early 2012) have become a major concern for commercial technologies based on this precious metal. In a sense, Pt can be viewed more as an industrial than as a precious metal. Since most of the current demand comes from vehicle catalytic converter makers, its price directly correlates with economic expansion [6].
Novel methods have avidly been sought to combine Pt with a more widely and promptly available metal to generate bimetallic nanocrystals (NCs) with structures shaped as alloys, dendrites, or core shells, to further reduce Pt loading in a catalyst [6–9].
Of the metals located in the vicinity of Pt in the periodic table, Pd is probably the best candidate for composing bimetallic NCs with Pt, as both exhibit the same face-centered cubic (fcc) structure and have nearly identical lattice constants (with a mismatch of only 0.77 %). Since Pd is much less expensive than Pt (the current price is only 40 % that of Pt), it makes more sense to reduce the cost of a Pd–Pt bimetallic catalyst by employing Pd as the base metal [6].
In a bimetallic Pt–Pd catalyst, the low compressive strain exerted by the weak lattice contraction causes a downshift in the d-band center, thereby reducing binding strength towards the adsorbed intermediates. This can diminish or enhance the catalytic activity, depending on whether adsorption or desorption of intermediates limits the reaction rate [6]. A small mismatch in the lattice constants of Pt and Pd, together with changes in Pt d-band properties caused by its coupling with Pd, makes Pd–Pt bimetallic NCs attractive catalysts for a variety of reactions, including ORR [6].
A Pd–Pt bimetallic nanodendrite is composed of a Pd NC at the core and branched arms of Pt at the periphery. These Pt arms, supported on a faceted Pd NC, are unique in that they generate a relatively large and highly accessible surface that can be particularly active towards a specific reaction [6].
Zhang et al. [10] reported that the electrochemical measurements of ORR activity for AuPd@Pt dendrimer-encapsulated nanoparticles (DENs) were in good agreement with the theoretical prediction that the activity peak for O binding should be slightly weaker than on a bulk Pt (1 1 1) surface. Sasaki et al. [11] developed a Pd9Au1 alloy core/Pt monolayer shell (Pd9Au1/PtML) electrocatalyst with increased stability—owing to the positive shift of Pd oxidation potential and lesser Pd–OH formation—and increased mass activity at 0.9 V. The Pd9Au1/PtML nanoparticles (NPs) exhibited improved stability, owing to Au-induced resistance to formation of Pd–OH and Pt–OH bonds under ORR conditions [12]. Zhang et al. [13], having synthesized Pt monolayer catalysts on Pd and Pd–Au hollow cores that were made using carbon-supported pulse-electrodeposited Ni NPs as sacrificial templates, claimed that total metal mass activity for ORR was enhanced, compared with both the Pt monolayer catalyst on solid Pd cores and solid pulse-deposited Pt NPs. Sasaki et al. [14] synthesized Pd9Au1 by wet impregnation of acetylene black with Pd and Au chlorides followed by chemical reduction with formic acid. PtML were deposited on a Pd–Au NP surface by galvanic displacement of a Cu monolayer. They claimed that the high stability of these NPs is caused by Au atoms segregating preferentially at defect sites in Pt monolayers, thus avoiding dissolution of Pd atoms through vacancies in these monolayers—Au atoms mend these defective sites. Adzic and co-workers [15, 16] synthesized a PtML by Cu underpotential deposition (UPD) on commercial Pd/C catalysts covered with a monolayer of Pd–Au alloy (Pd–Au alloys differed in atomic composition, depending on the catalyst employed) and attributed the enhanced ORR activity observed to PtML surface strain and d-band center shift induced by the alloy layer. Kuttiyiel et al. [17] produced Pd–Ni core shell NPs that were protected against decomposition in acid by Au atoms and activated for oxygen reduction with a Pt monolayer (PtMLAu10Pd40Ni50).
Bimetallic catalyst made of Pt on Pd produces a small mismatch in the lattice constants between Pt and Pd, in addition to changes in the d-band properties of Pt, caused by Pd coupling. This makes Pd–Pt bimetallic NCs attractive catalysts for ORR. A Pd–Pt bimetallic nanodendrite composed of a Pd NC at the core and branched peripheral Pt arms supported on a faceted Pd NC generates a large, highly accessible surface that might prove particularly active towards ORR. Finally, Pd9Au1/PtML NPs showed improved stability, owing to Au-induced resistance to formation of Pd–OH and Pt–OH bonds under ORR conditions.
Taking these aspects into account, the purposes of this study were (1) to synthesize NCs of a Pt or Pd or Pt–Pd alloy shell supported on a Au core, (2) to investigate the electrochemical-change profile of these catalysts using cyclic voltammetry (CV), and (3) to investigate their electrocatalytic activities and reaction mechanisms in ORR using hydrodynamic cyclic voltammetry (HCV) with a rotating ring-disk electrode (RRDE). TEM, HRTEM, HAADF-STEM, SEM, electron diffraction pattern, energy-dispersive X-ray (EDX) microanalysis, atomic absorption microanalysis, and XRD were employed to characterize the electrocatalysts.
Theoretical studies of systems that use combined Pt, Pd, and Au formulations suggest that a small, controlled amount of Au can improve the performance of PtPdAu thin films, retarding the dissolution of under-coordinated Pt atoms, possibly even enhancing their activity, making ternary PtPdAu systems interesting for ORR in proton-exchange membrane fuel cells [18]. We found that nanodendrites of Pt or Pt–Pd alloy supported on a Au core exhibit favorable traits in terms of catalysis and stability toward ORR.
Experimental
Reagents and instruments
The salts, precursor solutions, and solvents employed were HAuCl4·3H2O (Sigma-Aldrich), Brij 30 (Aldrich), PdCl2, NiCl2·6H2O, K2PtCl6, trisodium citrate, ascorbic acid, NaBH4, ethanol, and acetone (all Vetec).
Voltammetric measurements were carried out using a three-electrode glass cell with a working electrode consisting of a Teflon-embedded glassy carbon (GC)-disk/Au-ring rotating assembly (0.196 and 0.11 cm2 geometric areas, respectively) (Pine Research Instrumentation), with a collection efficiency of N = 0.26. The counter-electrode was a Pt plate (Degussa). A reversible hydrogen electrode (RHE) was employed as the reference electrode.
Electrode preparation
The GC-disk working electrode was sequentially polished with 2500-grit emery paper and alumina slurries (1 and 0.05 μm) and finally cleaned by sonication in Milli-Q water (Millipore), acetone, and 0.1 M HClO4 (Tedia) solution, for 5 min in each solvent. The Au ring underwent 200 cycles at 900 mV s−1 in the 0.05–1.7-V potential range (changing the solution whenever necessary to ensure a clean surface finish). Then, a uniform thin film was generated by dripping 30 μL of an aqueous solution of NCs at 1 mg mL−1 (loading, 152.7 μg cm−2) onto a GC disk surface and allowed to dry at room temperature. The modified electrode was placed in an electrochemical cell that contained a 0.1 M aqueous solution of HClO4. The solutions were saturated with N2 (5.0 purity) or O2 (4.0 purity), both from Air Liquide.
Sample preparation for TEM, HRTEM, SEM, and EDX measurements
For TEM, HRTEM, and EDX experiments, diluted NC aqueous solutions were dripped onto ultrathin carbon films supported by a lacey carbon film on a 400-mesh copper grid (Ted Pella). For SEM and EDX measurements, NC aqueous solutions were dripped onto GC plates (0.5 cm × 0.5 cm × 0.2 cm) (loading, 152.7 μg cm−2).
Apparatuses and measurements
CV and HCV, the electrochemical techniques employed, were run on an AFCBP1 bipotentiostat coupled to an MSRX speed controller, both from Pine Research Instrumentation.
A PGSTAT128N potentiostat/galvanostat (Autolab) equipped with a FRA2.X module was used in the electrochemical impedance spectroscopy (EIS) experiments, conducted at a fixed potential—namely, at an open-circuit potential (OCP) of around 1.06 V (on average) in the presence of a 0.1 M aqueous solution of HClO4, with potential perturbation of 25 mV (rms) within a frequency range of 10 mHz to 100 kHz. Care was taken to ensure that AC impedance data corresponded to the interfaces being investigated at high frequencies, as described in reference 19. Each HCV curve was corrected for ohmic drop resistance measured and determined from the fitted high-frequency intercept measured using EIS. The average value of measured ohmic drop resistance in 0.1 M HClO4 ranged from 26 to 35 Ω.
NC micro- and nanostructures were characterized by both TEM and SEM. TEM was performed on a JEM 2100F apparatus (JEOL) equipped with a scanning transmission electron microscope (STEM) and EDX (Thermo Noram system), operating at 200 kV. SEM was performed on a JEOL JSM-6380LV field-emission scanning electron microscope (an accelerating voltage of 30 kV was applied) equipped with an EDX system (Thermo Noran System Six) to microanalyze Au, Pd, Pt, and Ni ratios in the NCs. The atomic compositions reported represent the average of five measurements of the same sample. Also, NC compositions were evaluated by atomic absorption using an Analyst 200 apparatus (Perkin Elmer) comparable to an EDX microanalyzer. NC structural characterization was based on XRD measurements performed with a XRD 6000 diffractometer (Shimadzu) using Cu Kα radiation (λ = 0.15406 nm). The diffractograms were recorded at 2θ in the 10°–80° range.
Cyclic voltammograms were normalized to the electrochemically active surface area (ECSA). For NCs involving Pt, this was done by integrating charges in the region of hydrogen desorption; when NCs involved Pd (absence of Pt), charge integration encompassed the region of oxide reduction. Charge density was taken as 210 μC cm−2 for desorption of a hydrogen monolayer from the Pt surface and as 420 μC cm−2 for reduction of an oxygen monolayer covering the Pd surface, respectively [20]. We kept the same CV potential scan limits to NCs involving Pd (absence of Pt) as NCs involving Pt to facilitate response comparisons even knowing the controversy reported by Grdeń et al. [21] that the completion of formation of the first monolayer (ML) of Pd2+ oxide occurs in the 1.45–1.50 V vs. RHE potential range contrary to reports suggesting that the growth of the first ML of Pd2+ oxide is completed at 1.1–1.3 V vs. RHE (especially in basic electrolytes). An ECSA value of 0.43 cm2 for the GC electrode was determined as described by Fortunato et al. [22]. Briefly, cyclic voltammogram responses for a bare GC electrode in a N2-saturated 0.1 M HClO4 solution containing a total 2 mM concentration of K3[Fe(CN)6]/K4[Fe(CN)6] (1:1 molar ratio) were used to determine its ECSA value.
NC synthesis
Synthesis A (Scheme 1)
A sol containing Au seeds with a diameter of about 28 nm was synthesized according to Frens’ method [23]. In order to add Pd monolayers onto the Au surface, 45 mL of sol containing Au seeds was mixed with 0.565 mL of 0.08 M aqueous PdCl2 and then cooled to 0 °C in an ice bath [24]. Subsequently, 1 mL of 0.48 M aqueous ascorbic acid (AA) was quickly added to the mixture while stirring. The mixture was then stirred for 3 h at 0 °C to ensure complete reduction of PdCl2 [24]. The red brown color of the mixture turned dark brown, indicating formation of Pd@Au NCs [24]. In order to add Pt clusters onto the Pd surface, the above mixture was heated to 80 °C, added with 0.565 mL of 0.08 M aqueous K2PtCl6 dropwise [24], and heated to 80 °C for several minutes [25–27], after which 1 mL of 0.24 M aqueous AA was added (through a dripper, under vigorous stirring), and the mixture was allowed to react for another 3 h [24–26] at 80 °C to ensure complete reduction of K2PtCl6 [24]. Finally, the Pt–Pd@Au NC solution was centrifuged for 30 min, washed and centrifuged five times with Millipore water at 4500 rpm, and dried at 40 °C. Pd@Au and Pt@Au NCs were obtained following the above procedure, but skipping the step corresponding to the absent metal. To produce Pt and Pd NCs, only the steps corresponding to each metal were carried out (in 45 mL of water), followed by centrifugation–wash–centrifugation.

Synthesis procedures
Synthesis B (Scheme 1)
Ni–Au NCs were chemically prepared through a redox-transmetalation method in reverse microemulsion [28]. The reaction was carried out under stirring and N2 protection at room temperature. Brij 30 and ethanol were used as the surfactant and the oil phase, respectively. In the reverse microemulsion, water droplet size, which constrains NC growth, was tentatively controlled by adjusting the value of Ω (the water-to-surfactant molar ratio) [28]. Ni cores were prepared by reducing Ni2+ with NaBH4, and an Ω value of 7 was selected to prepare the initial Ni nanoparticles [28]. To begin, 1.8 mL of a NiCl2·6H2O (0.049 M) aqueous solution was added to a 21.6 mL mixture composed of 5.4 mL of Brij 30 and 16.2 mL of ethanol to form the first reverse microemulsion solution. The second reverse microemulsion solution, containing 0.6 mL of NaBH4 (0.45 M) and prepared at an identical water-to-surfactant molar ratio as the first, was then added dropwise. The solution immediately turned black, owing to formation of nickel nanoparticles. To ensure thorough reduction of NiCl2, we designed the NaBH4-to-NiCl2 molar ratio to be 2 [28] and kept the reaction solution under N2 protection for 12 h, a period also deemed sufficient to ensure the spontaneous decomposition of excess NaBH4, a compound whose persistence in the solution would disturb the sequential redox-transmetalation reaction [28]. Formation of Au shells on these Ni cores is driven by an in situ redox-transmetalation reaction between Ni atoms (on the surface of Ni NPs) and AuCl4 − ions, without any additional reducing agent [28]. The reverse microemulsion solution (0.9 mL) containing 0.675 mL of HAuCl4 (0.1 M) + 0.225 mL of water was slowly dripped onto the solution of the initial Ni NPs. The Ω value of the HAuCl4 solution was maintained at 7 (1.425 mL of Brij 30 and 4.275 mL of ethanol). The Ni-to-Au molar ratio was kept at 2 to ensure formation of Au shells of proper thickness, so as to prevent overeroding the initial Ni NPs. The solution was maintained under N2 protection for another 12 h to ensure completion of the redox-transmetalation process. In order to add Pt clusters to the Au surface, the above solution was heated to 80 °C in an open environment and 0.6 mL of 0.08 M K2PtCl6 ethanol solution was dripped onto it [24], followed by heating to 80 °C for several minutes [25–27]. Subsequently, 1 mL of 0.254 M AA ethanol solution was added (through a dripper, under vigorous stirring), and the mixture was allowed to react for another 3 h [24–27] at 80 °C to ensure complete reduction of K2PtCl6 [24]. Finally, the now designated Pt@Au′ NC solution was centrifuged for 30 min, washed and centrifuged five times with Millipore water, five times with acetone, and five times with Millipore water at 4500 rpm, and finally dried at 40 °C.
Results and discussion
Comparing the NCs generated
The structures of the NCs produced can be further understood considering the TEM and HRTEM images and the initial and EDX mass percentage values depicted in Table 1, and SEM images depicted in Supporting Information Table S1.
Pt NCs are heterogeneous, with spherical, triangular, squared, pentagonal, hexagonal, heptagonal, and rod-like shapes (see TEM images in Table 1), have large-sized structures (80 nm on average), and exhibit a nanodendritic covering pattern (see HRTEM image in Table 1). Similar shapes are observed in other NCs, with Pt@Au NCs slightly more rod-shaped, while Pt–Pd@Au NCs have slightly less defined shapes. However, nanodendritic coverings are also present on the surface of these NCs (see TEM and HRTEM images in Table 1). The shapes of GC-supported NCs are relatively similar in the SEM and TEM images (Supporting Information Table S1 and Table 1), despite the different resolutions employed for each technique. No considerable differences were observed between initial metal mass percentages before and after NC synthesis, with the exception of Pt@Au′ NCs, given that no metallic Ni was detected and the particles are rich in metallic Au. Au enrichment occurs to a lower extent in Pt@Au NCs (Table 1). In the TEM images, NCs exhibit similarities with TEM images from elsewhere for a Pt–npAu composite [1], Pd–Pt bimetallic nanodendrites [6], Pt–Pd alloy icosahedra [29], Au@Pd NCs [30], Au@Pd core shell NPs [31], and Au@Pd core shell NCs [32]. Our TEM images of Pt–Pd@Au NCs bear some similarity with a TEM image of 55-nm Au@Pd@Pt NPs with two Pd monolayers and 20 Pt monolayers synthesized by Fang et al. [24], while our SEM images of Pt@Au NCs are somewhat similar to SEM images of 55-nm Au@0.7–1.4 nm Pt NPs obtained by Tian and co-workers [25–27]. However, it was not possible to detect similarities between our TEM images of Pt@Au′ NCs and those of Ni@Au NPs synthesized by Chen et al. [28].
EDX mapping images in Table 2 clearly reveal that the base of NCs is constituted of metallic Au nanostructures, covered with a Pt nanodendrite surface in the case of Pt@Au NCs. For Pt–Pd@Au NCs, the base is constituted of metallic Au nanostructures covered with a Pt–Pd alloy nanodendrite surface. Pt@Au′ NCs have a base constituted of metallic Au nanostructures covered with a Pt nanodendrite surface; in this case, these Au nanostructures result from Ni overerosion during the redox-transmetalation process. These EDX mapping images corroborate the EDX mass percentages shown in Table 1.
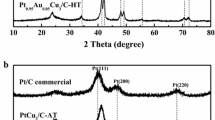
The XRD patterns shown in Fig. 1 exhibit peaks characteristic of face-centered cubic (fcc) crystalline Au, Pt, and Pd. For Pt@Au NCs, diffraction peaks corresponding to Au (1 1 1), (2 0 0), (2 2 0), and (3 1 1) planes and shoulders corresponding to Pt (1 1 1), (2 0 0), and (2 2 0) planes are observed at 2θ values of 38.3° and 39.5°, 44.5° and 46.0°, and 64.8° and 67.3° (for Au and Pt, respectively) and 2θ = 77.7° (for Au alone) (PCPDF 040784 and 652868 for Au and Pt, respectively). For Pt–Pd@Au, diffraction peaks corresponding to Au and Pt and Pd (1 1 1), (2 0 0), (2 2 0) planes and to Au (3 1 1) plane are observed at 2θ values of 38.3° and 39.8°, 44.5° and 46.6°, and 64.8° and 68.0° (in each pair, the first value for Au and the second value for Pt and Pd) and 2θ = 77.7° (for Au alone) (PCPDF 040784, 652868, and 894897 for Au, Pt, and Pd, respectively). For Pt@Au′, diffraction peaks corresponding to Au (1 1 1), (2 0 0), (2 2 0), and (3 1 1) planes are observed at 2θ values of 38.3°, 44.5°, 64.7°, and 77.7°, respectively, while a mild shoulder (see insets to Fig. 1) corresponding to Pt (1 1 1) plane is barely visible at 2θ = 40°, and no peaks or shoulders corresponding to fcc crystalline Ni were identified (2θ values of 44.5°, 51.8°, and 76.4° for Ni (1 1 1), (2 0 0), and (2 2 0) planes, respectively; PCPDF 701849) (see insets to Fig. 1).

XRD patterns for Pt@Au, Pt–Pd@Au, and Pt@Au′ NCs
A noteworthy feature was that the peak and shoulder intensities depicted in Fig. 1 directly correlated with metal mass percentages found after NC synthesis, shown in Table 1. Au (1 1 1) peak was used to calculate average crystallite sizes (ACSs), applying Scherrer’s formula [33]: ACS = (0.94λ)/(βcosθ), where 0.94 is an approximate constant for sphere samples, λ is the X-ray wavelength, β is the half-width of the diffraction peak (in radians) in relation to Si (used as a standard), and θ is the diffraction angle. The values thus calculated were 19 nm (Pt@Au NCs), 27 nm (Pt–Pd@Au NCs), and 37 nm (Pt@Au′ NCs), lower therefore than the sizes reported in Table 1. (It should be borne in mind, however, that calculations based on Scherrer’s formula considered Au (1 1 1) fcc crystallites as spherical, when in fact ACS values should apply.)
Additional information about fcc crystalline Au, Pt, and Pd can be found in Table 3, which shows electron diffraction patterns of the NCs synthesized.
For Pt NCs, the diffraction image in Table 3 clearly shows the fcc crystalline pattern of the Pt [1 1 2] zone axis, with main planes (1 1 1), at a distance of 0.229 nm, and (2 2 0), at a distance of 0.140 nm. For Pt@Au NCs, the central part of the electron diffraction image shows the fcc crystalline pattern of the Au [1 1 0] zone axis face, with main planes (1 1 1), at a distance of 0.238 nm, and (0 0 2), at a distance of 0.205 nm, but no rings indicating that the Pt nanodendrites at the edges are oriented towards and attached onto the Au surface. For Pt–Pd@Au NCs (Table 3), the central part of the electron diffraction image shows an fcc crystalline pattern of Au at one of the Au zone axes (the diffraction pattern comprises the entire image), with planar Au distances of 0.234 nm and 0.143 nm. Similarly, Pt–Pd alloy nanodendrites at the edges are oriented towards and attached onto the Au surface, yet to a lesser degree than Pt nanodendrites on Pt@Au NC surfaces. A different crystalline pattern is observed for Pt@Au′ NCs, since the electron diffraction image in Table 3 differs from those depicting other NCs containing Au (see also the EDX mass percentages shown in Table 1); also, a poorly organized crystalline pattern occurs at the edges of the electron diffraction image depicting Pt nanodendrites.
Electrochemical profile
Figure 2 shows electrochemical profiles for bare and NC-modified GC electrodes recorded in a N2-saturated 0.1 M aqueous solution of HClO4.

Cyclic voltammograms for bare and NC-modified GC electrodes in N2-saturated 0.1 M HClO4. Scan rate, 50 mV s−1. Scans started at 1.2 V
The results show the typical behavior regarding the hydrogen and oxide regions for Pt and Pd surfaces, consistent with previous reports [22, 34]. For bare GC, no faradaic responses were detected [22].
A noteworthy feature is that GC electrodes modified with NCs devoid of Pt (see green and cyan curves, Fig. 2) exhibited higher current densities than those modified with NCs containing Pt, owing to the smaller ECSA values of the former.
Evaluation of the electrocatalytic activity of modified GC electrodes towards ORR
Figure 3 shows the oxygen reduction polarization curves for bare and NC-modified GC electrodes in an O2-saturated 0.1 M aqueous solution of HClO4. The HCV curves were obtained with subtraction of background HCV curves for bare and GC electrodes modified with NCs in an N2-saturated 0.1 M aqueous solution of HClO4. Current densities were calculated per geometric area of bare GC electrode.

Hydrodynamic cyclic voltammograms for bare and NC-modified GC electrodes in O2-saturated 0.1 M HClO4. ω = 1600 rpm; scan rate, 10 mV s−1. Scans started at 0.05 V
On Pt–Pd@Au, Pt@Au, Pt@Au′ NCs supported on GC electrodes (see dark yellow, blue, and magenta curves, Fig. 3), the positive-direction potential scan revealed that O2 reduction started in close vicinity to the maximum current density (j d ), with a very small increase in current densities (in absolute value) up to about 0.31 V, and was responsible for producing small amounts of H2O2 (0.5 % for Pt@Au and Pt@Au′ NCs). (Note the small currents decreasing until near 0.31 V in the black and green curves, Fig. S1, Supporting Information; see Supporting Information also for Equations S1 and S2, used to calculate the percentage of H2O2 formation, X H2O2) [34].
From 0.31 to 0.75 V (see dark yellow, blue, and magenta curves, Fig. 3), O2 reduction peaked (the j d region) and H2O2 synthesis declined above 0.31 V (to 0.3 % for Pt@Au′ NCs—see Fig. S1, Supporting Information, including negative-direction scan). Negligible H2O2 production (0.06 %) was detected for Pt–Pd@Au NCs (see red curve in Supporting Information Fig. S1, including negative-direction scan). Proceeding in the positive direction, a potential region was found that lacked O2 reduction (zero current densities), close to 1.05 V (the Pt surface was completely covered with PtO x ) [22, 34]. Reversing the potential scan direction caused current densities related to O2 reduction to be 0 until the potential reached about 0.98 V (the potential at which PtO x reduction began) [22, 34]. Proceeding towards more-negative potentials, current densities increased throughout the kinetic region and reached the potential region in which j d prevails. For these modified electrodes, E 1/2 (the potential corresponding to half the value of j d ) was about 0.93 V in the positive-direction scan (the highest value obtained herein and close to the value reported by Venarusso et al. [34]) and about 0.86 V in the negative-direction scan. This 70-mV difference between E 1/2 values supports the assumption that the absence or presence of PtO x influences ORR catalysis at these modified electrodes [22, 34].
The second highest sequence of E 1/2 values was obtained for the GC electrode modified with Pt NCs (see red curve, Fig. 3). Smaller E 1/2 values were obtained for GC electrodes modified with Pd@Au and Pd NCs.
Table 4 (see also Table S2, Supporting Information) shows calculated specific activity values (SA [22, 34, 36, 37]; see Equation S3, Supporting Information) at potentials close to E 1/2. Calculated ECSA values for modified GC electrodes are shown in Table 4 and Table S2, Supporting Information. Particularly high SA (at 0.90 V, close to E 1/2) and ECSA values were found for Pt@Au (1.22 mA cm−2) and Pt (5.0 cm2), respectively. The positive catalytic influence exerted by Au on activation barriers and reaction energies for ORR [38] after Pt deposition under the Au surface were previously described by our group [34], as were the positive catalytic influence of Pd on activation barriers and reaction energies for ORR [38] after Pt and Pd deposition under the GC surface [22] (see the highly positive E 1/2, SA, and ECSA values for Pt@Au, Pt@Au′, and Pt–Pd@Au in Table S2).
Low platinum group metal (PGM) loadings of 27.5, 85.5, and 50.4 μg cm−2 (Table 4) resulted in specific ECSA values of 69, 25, and 34 m2 g−1 and mass-specific activity values of 0.47, 0.30, and 0.41 mA μg−1 at 0.9 V (see Supporting Information for Equations S4 and S5, used to calculate PGM specific ECSA and PGM mass-specific activity) for GC electrodes modified with Pt@Au′, Pt–Pd@Au, and Pt@Au NCs, respectively. These values were better, in some cases, than those reported elsewhere (Table 4), not only indicating very good electrocatalytic activity but also suggesting economic viability, by requiring less than 100 μg of Au and 50 μg of finely dispersed Pt per square centimeter to generate very thin films [1].
Even considering the total metal loading (Table S3), the values of MSA are higher for Pt@Au′, Pt@Au, and Pt–Pd@Au NCs in comparison with Pt NCs—and comparable to state-of-the art Pt/C catalysts [35] (Table 4)—in addition to approaching their specific ECSA values (ECSA values are already similar, Table 4), reinforcing the view that the main influences promoting effective ORR catalysis by NCs are compositional and surface structural (nanodendrites) features, instead of NC sizes.
In summary, the good electrocatalytic behavior towards ORR exhibited by Pt or Pt–Pd alloy electrocatalysts synthesized over a Au core (including Au cores produced from redox-transmetalation of Ni cores)—namely, Pt–Pd@Au, Pt@Au, and Pt@Au′ NCs—may be explained by (a) the presence of Pt or Pt–Pd alloy on top of these NCs and (b) the occurrence of Pt or Pt–Pd alloy dendrimer-encapsulated NC structures [10]. Pinto and Maia [39] conducted a theoretical study of the adsorption of atomic oxygen onto a surface alloy of palladium and platinum deposited on a gold substrate, based on our prior experimental work [34]. The results show that oxygen adsorption is more stable on a hollow fcc site of this surface, where the binding energy is in the order of −2.3 eV. The oxygen atom has lower affinity for Pd and adsorption does not take place on top sites (higher energy and greater distance) [39]. This is a consequence of the interaction between the oxygen orbital and the d-band of Pd and Pt [39]. The valence orbital shifts toward the Fermi level, and its structure changes as a result of coupling between Pd and Pt d-bands and the p orbital of oxygen [39].
ORR mechanism on the surface of NC-modified GC electrodes was determined from hydrodynamic linear potential scan curves recorded at different rotation rates (Fig. S2a, Supporting Information). According to the Koutecký–Levich plots [40] (Fig. S2b, Supporting Information), the number of electrons transferred (n) per oxygen molecule from NC catalysts was calculated (Equation S6, Supporting Information) to be in average 4 (see Table S4, Supporting Information). These results suggest that a four-electron process is the dominating pathway for the oxygen reduction resulting in water at the NC-modified GC electrodes, as observed in previous works [22, 34]. This is corroborated by the low production of H2O2 observed, of 0.5 % at most.
The values calculated (see Table S5, Supporting Information) from Tafel plots [40] (E vs. log(I/(I d − I), Fig. S2c, Supporting Information) were close to the expected values of −60 and −120 mV dec−1 for the low- and high-current regions reported to occur during ORR on bulk Pt [22, 34, 41] and Pd [22, 34, 42].
Electrocatalytic stability tests
In addition to the assessment of catalytic activity, we carried out an accelerated durability test to evaluate the long-term stability of the GC electrodes modified with Pt@Au and Pt–Pd@Au NCs (Fig. 4). The test was conducted by applying a continuous potential cycling between 0.6 and 1.0 V at 50 mV s−1 in an O2-saturated 0.1 M aqueous solution of HClO4, based on our previous investigations [22, 34] and drawing on other published reports [43, 44].

Hydrodynamic cyclic voltammograms for GC electrodes modified with a Pt@Au and b Pt–Pd@Au NCs, before and after stability test (10,000 cycles between 0.6 and 1.0 V at 50 mV s−1) in O2-saturated 0.1 M HClO4. Ω = 1600 rpm; scan rate, 10 mV s−1. Scans started at 0.05 V
Two of the catalysts found to have superior catalytic activity after 10,000 potential scans (GC electrodes modified with Pt@Au and Pt–Pd@Au NCs, Fig. 4) exhibited no detectable differences in comparison with CVs before 10,000 cycles (Fig. S3, Supporting Information). However, after 10,000 cycles, ECSA values decreased to 2.1 and 2.8 cm2 (38 and 33 % losses, from 3.3 and 4.2 cm2, respectively; see Table 4). Mass specific activity, specific ECSA, and SA correction for ECSA values of 2.1 and 2.8 cm2 were 0.36 and 0.26 mA μg−1, 21 and 17 m2 g−1, and 1.71 and 1.54 mA cm−2, respectively, at 0.90 V, which were lower than specific ECSA values. However, these decreases can be considered negligible for mass-specific activity, whereas a high increase was observed in comparison with SA values before 10,000 potential scans (Table 4).
Table 5 and Supporting Information Table S6 show representative TEM, HRTEM, HAADF-STEM and EDX mapping images, and electron diffraction patterns for Pt@Au and Pt–Pd@Au NCs after 10,000 potential scans.
After 10,000 potential scans, shape changes and electron diffraction patterns for NCs were clearly absent, while NC edges appeared smoother. Pt or Pt–Pd alloy nanodendrite structures at NC edges became less pronounced, without dissolution of inner metallic nanoparticles such as Au (see EDX mapping in Table 5), which may explain the decreased ECSA and specific ECSA values obtained after 10,000 potential scans.
A positive aspect is that NC shape and the preservation of a large number of nanodendrites at the NC surfaces were sufficient to keep mass-specific activity quite unchanged and increase SA after 10,000 potential scans (Table 4).
These decreases in ECSA and specific ECSA values may be explained by Ostwald ripening [22, 34, 45, 46]—i.e., dissolution and redeposition of Pt or Pt–Pd alloy nanodendrites at NC edges [22, 34, 45, 46]. We found that Pt or Pt–Pd alloy nanodendrite dissolution and redeposition have no deleterious effects on mass-specific activity and SA of the NC catalysts investigated. Au cores induced resistance to formation of Au–OH [11, 12], keeping the heterogeneous NC structure intact, while, to a lesser degree, promoting Pt–OH or Pd–OH formation, thus smoothing Pt or Pt–Pd alloy nanodendrites.
Loss of specific ECSA was, on average, similar to that reported in the literature (Table 4). In terms of mass-specific activity and SA, our values were comparatively very good.
In summary, at least three factors may be responsible for the good electrocatalytic activities of our Pt NC catalysts: (1) presence of Pt or Pt–Pd alloy on top of NCs; (2) presence of NC structures containing encapsulated Pt or Pt–Pd alloy dendrimers; and (3) metal-dissolution/deposition processes that are involved in the stability of NC electrocatalysts, including variations in the elemental identity of admetals and substrates and changes in local geometric structure [47, 48]. After the stability tests, the NC electrocatalysts that underwent some degree of Pt or Pt–Pd alloy nanodendrite dissolution/deposition preserved good mass-specific activity and SA values, given that Au cores kept the heterogeneous NC structure investigated intact. However, NC electrocatalysts containing only Pd or Pd@Au failed to demonstrate good electrocatalytic behavior towards ORR.
Conclusions
Pt or Pt–Pd alloy electrocatalysts synthesized over a Au core (including Au cores produced from redox-transmetalation of Ni cores)—namely, Pt–Pd@Au, Pt@Au, and Pt@Au′ NCs—effectively catalyzed ORR, with high performance in terms of Pt mass-specific activity and SA. These results showed that these Pt-containing NC catalysts exhibited high electrocatalytic activity towards ORR (in terms of ECSA and specific ECSA values at E 1/2 ≈ 0.93 V). GC electrodes modified with Pt–Pd@Au, Pt@Au, and Pt@Au′ NCs, which had the highest electrocatalytic activity, contained heterogeneous NCs with spherical, triangular, squared, pentagonal, hexagonal, heptagonal, and rod-like shapes, with large (about 80 nm in several cases), well-defined crystalline structures, and exhibited a nanodendritic Pt or Pt–Pd alloy covering pattern at the NC surface, which underwent structural changes in Pt or Pt–Pd alloy nanodendrites on the NC edges, due to dissolution/deposition after 10,000 potential cycles (stability test), with decreased ECSA and specific ECSA values, stable mass-specific activity, and increased SA values, features that suggest their utility for use in acid fuel cells. Electrocatalysts containing Pd NCs alone or Pd@Au NCs failed to demonstrate good electrocatalytic behavior towards ORR.
References
Wittstock A, Wichmann A, Bäumer M (2012) Nanoporous gold as a platform for a building block catalyst. ACS Catal 2:2199–2215
Gong K, Park J, Su D, Adzic RR (2014) Metalizing carbon nanotubes with Pd–Pt core–shell nanowires enhances electrocatalytic activity and stability in the oxygen reduction reaction. J Solid State Electrochem 18:1171–1179
Zhang G-R, Zhao D, Feng Y-Y, Zhang B, Su DS, Liu G, Xu B-Q (2012) Catalytic Pt-on-Au nanostructures: why Pt becomes more active on smaller Au particles. ACS Nano 6:2226–2236
Fennell J, He D, Tanyi AM, Logsdail AJ, Johnston RL, Li ZY, Horswell SL (2013) A selective blocking method to control the overgrowth of Pt on Au nanorods. J Am Chem Soc 135:6554–6561
Hartl K, Mayrhofer KJJ, Lopez M, Goia D, Arenz M (2010) AuPt core–shell nanocatalysts with bulk Pt activity. Electrochem Commun 12:1487–1489
Zhang H, Jin M, Xia Y (2012) Enhancing the catalytic and electrocatalytic properties of Pt-based catalysts by forming bimetallic nanocrystals with Pd. Chem Soc Rev 41:8035–8049
Li C, Yamauchi Y (2013) Facile solution synthesis of Ag@Pt core–shell nanoparticles with dendritic Pt shells. Phys Chem Chem Phys 15:3490–3496
Wang L, Yamauchi Y (2010) Controlled aqueous solution synthesis of platinum–palladium alloy nanodendrites with various compositions using amphiphilic triblock copolymers. Chem Asian J 5:2493–2498
Ataee-Esfahani H, Liu J, Hu M, Miyamoto N, Tominaka S, Wu KCW, Yamauchi Y (2013) Mesoporous metallic cells: design of uniformly sized hollow mesoporous Pt–Ru particles with tunable shell thicknesses. Small 9:1047–1051
Zhang L, Iyyamperumal R, Yancey DF, Crooks RM, Henkelman G (2013) Design of Pt-shell nanoparticles with alloy cores for the oxygen reduction reaction. ACS Nano 7:9168–9172
Sasaki K, Naohara H, Cai Y, Choi YM, Liu P, Vukmirovic MB, Wang JX, Adzic RR (2010) Core-protected platinum monolayer shell high-stability electrocatalysts for fuel-cell cathodes. Angew Chem Int Ed 49:8602–8607
Guo S, Zhang S, Sun S (2013) Tuning nanoparticle catalysis for the oxygen reduction reaction. Angew Chem Int Ed 52:2–21
Zhang Y, Ma C, Zhu Y, Si R, Cai Y, Wang JX, Adzic RR (2013) Hollow core supported Pt monolayer catalysts for oxygen reduction. Catal Today 202:50–54
Sasaki K, Naohara H, Choi YM, Cai Y, Chen W-F, Liu P, Adzic RR (2012) Highly stable Pt monolayer on PdAu nanoparticle electrocatalysts for the oxygen reduction reaction. Nat Commun 3:1115. doi:10.1038/ncomms2124
Xing Y, Cai Y, Vukmirovic MB, Zhou W-P, Karan H, Wang JX, Adzic RR (2010) Enhancing oxygen reduction reaction activity via Pd-Au alloy sublayer mediation of Pt monolayer electrocatalysts. J Phys Chem Lett 1:3238–3242
Cai Y, Adzic RR (2011) Platinum monolayer electrocatalysts for the oxygen reduction reaction: improvements induced by surface and subsurface modifications of cores. Adv Phys Chem Article ID 530397. doi:10.1155/2011/530397
Kuttiyiel KA, Sasaki K, Su D, Vukmirovic MB, Marinkovic NS, Adzic RR (2013) Pt monolayer on Au-stabilized PdNi core–shell nanoparticles for oxygen reduction reaction. Electrochim Acta 110:267–272
Tripkovic V, Hansen HA, Rossmeisl J, Vegge T (2015) First principles investigation of the activity of thin film Pt, Pd and Au surface alloys for oxygen reduction. Phys Chem Chem Phys 17:11647–11657
Venarusso LB, Tammeveski K, Maia G (2011) Versatile charge transfer through anthraquinone films for electrochemical sensing applications. Electrochim Acta 56:8926–8933
Angerstein-Kozlowska H (1984) Surfaces, cells, and solutions for kinetics studies. In: Yearger E, Bockris JO’M, Conway BE, Sarangapani S (eds) Comprehensive treatise of electrochemistry, vol 9. Plenum, New York, pp 15
Grdeń M, Łukaszewski M, Jerkiewicz G, Czerwiński A (2008) Electrochemical behaviour of palladium electrode: oxidation, electrodissolution and ionic adsorption. Electrochim Acta 53:7583–7598
Fortunato GV, Venarusso LB, Maia G (2014) Large platinum structures as promising catalysts for the oxygen-reduction reaction. Chem Electro Chem 1:625–636
Frens G (1973) Controlled nucleation for the regulation of the particle size in monodisperse gold suspensions. Nat Phys Sci 241:20–22
Fang P-P, Duan S, Lin X-D, Anema JR, Li J-F, Buriez O, Ding Y, Fan F-R, Wu D-Y, Ren B, Wang ZL, Amatore C, Tian Z-Q (2011) Tailoring Au-core Pd-shell Pt-cluster nanoparticles for enhanced electrocatalytic activity. Chem Sci 2:531–539
Li J-F, Yang Z-L, Ren B, Liu G-K, Fang P-P, Jiang Y-X, Wu D-Y, Tian Z-Q (2006) Surface-enhanced Raman spectroscopy using gold-core platinum-shell nanoparticle film electrodes: toward a versatile vibrational strategy for electrochemical interfaces. Langmuir 22:10372–10379
Zhang P, Chen Y-X, Cai J, Liang S-Z, Li J-F, Wang A, Ren B, Tian Z-Q (2009) An electrochemical in situ surface-enhanced Raman spectroscopic study of carbon monoxide chemisorption at a gold core–platinum shell nanoparticle electrode with a flow cell. J Phys Chem C 113:17518–17526
Zhang P, Cai J, Chen Y-X, Tang Z-Q, Chen D, Yang JL, Wu D-Y, Ren B, Tian Z-Q (2010) Potential-dependent chemisorption of carbon monoxide at a gold core-platinum shell nanoparticle electrode: a combined study by electrochemical in situ surface-enhanced Raman spectroscopy and density functional theory. J Phys Chem C 114:403–411
Chen D, Li J, Shi C, Du X, Zhao N, Sheng J, Liu S (2007) Properties of core-shell Ni-Au nanoparticles synthesized through a redox-transmetalation method in reverse microemulsion. Chem Mater 19:3399–3405
Gu J, Zhang Y-W, Tao F (2012) Shape control of bimetallic nanocatalysts through well-designed colloidal chemistry approaches. Chem Soc Rev 41:8050–8065
Kim D, Lee YW, Lee SB, Han SW (2012) Convex polyhedral Au@Pd core–shell nanocrystals with high-index facets. Angew Chem 124:163–167
Song HM, Anjum DH, Sougrat R, Hedhili MN, Khashab NM (2012) Hollow Au@Pd and Au@Pt core–shell nanoparticles as electrocatalysts for ethanol oxidation reactions. J Mater Chem 22:25003–25010
Zhang L-F, Zhang C-Y (2013) Dodecahedral Au@Pd nanocrystals with high-index facets and excellent electrocatalytic activity and highly efficient surface-enhanced Raman scattering enhancement. Nanoscale 5:6074–6080
Cullity BD (1956) Elements of X-ray diffraction. Addison-Wesley Publishing Company, Inc., Reading, Massachusetts
Venarusso LB, Sato RH, Fiorito PA, Maia G (2013) Platinum systems electrodeposited in the presence of iron or palladium on a gold surface effectively catalyze oxygen reduction reaction. J Phys Chem C 117:7540–7551
Gasteiger HA, Kocha SS, Sompalli B, Wagner FT (2005) Activity benchmarks and requirements for Pt, Pt-alloys, and non-Pt oxygen reduction catalysts for PEMFCs. Appl Catal B: Env. 56:9–35
Garsany Y, Baturina OA, Swider-Lyons KE, Kocha SS (2010) Experimental methods for quantifying the activity of platinum electrocatalysts for the oxygen reduction reaction. Anal Chem 82:6321–6328
Debe MK (2012) Electrocatalyst approaches and challenges for automotive fuel cells. Nature 486:43–51
Bligaard T, Nørskov JK (2008) Heterogeneous catalysis. In: Nilsson A, Pettersson LGM, Nørskov JK (eds) Chemical bonding at surfaces and interfaces. Elsevier, Amsterdam, p. 255
Pinto LMC, Maia G (2015) Oxygen adsorption on PdPt/Au(111)–DFT calculations. J Phys Chem C 119:8213–8216
Bard AJ, Faulkner LR (2001) Electrochemical methods: fundamentals and applications. John Wiley & Sons, New York
Sepa DB, Vojnovic MV, Damjanovic A (1981) Reaction intermediates as a controlling factor in the kinetics and mechanism of oxygen reduction at platinum electrodes. Electrochim Acta 26:781–793
Vracar LM, Sepa DB, Damjanovic A (1986) Palladium electrode in oxygen-saturated aqueous solutions—reduction of oxygen in the activation-controlled region. J Electrochem Soc 133:1835–1839
Lim B, Jiang M, Camargo PHC, Cho EC, Tao J, Lu X, Zhu Y, Xia Y (2009) Pd-Pt bimetallic nanodendrites with high activity for oxygen reduction. Science 324:1302–1305
Wu G, More KL, Johnston CM, Zelenay P (2011) High-performance electrocatalysts for oxygen reduction derived from polyaniline, iron, and cobalt. Science 332:443–447
Xu Y, Shao M, Mavrikakis M, Adzic RR (2009) Recent developments in the electrocatalysis of the O2 reduction reaction. In: Koper MTM (ed) Fuel cell catalysis: a surface science approach. John Wiley & Sons, New Jersey, p. 271
Sasaki K, Shao M, Adzic R (2009) Dissolution and stabilization of platinum in oxygen cathodes. In: Büchi FN, Inaba M, Schmidt TJ (eds) Polymer electrolyte fuel cell durability. Springer, New York, pp. 7–27
Greeley J, Markovic NM (2012) The road from animal electricity to green energy: combining experiment and theory in electrocatalysis. Energy Environ Sci 5:9246–9256
Greeley J (2010) Structural effects on trends in the deposition and dissolution of metal-supported metal adstructures. Electrochim Acta 55:5545–5560
Acknowledgments
The authors wish to thank LabMic UFG for the microscopy analyses and LNNano for the TEM facilities. Thanks are also given to CNPq (grants 301403/2011-2, 473991/2012-8, 405695/2013-6, 303759/2014-3, and 442268/2014-9) and Fundect-MS (grants 23/200.583/2012, 23/200.735/2012, and 23/200.246/2014) for their financial support. L.B.V. thanks CAPES for the fellowship.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(DOCX 24581 kb)
Rights and permissions
About this article

Cite this article
Venarusso, L.B., Bettini, J. & Maia, G. Catalysts for oxygen reduction reaction based on nanocrystals of a Pt or Pt–Pd alloy shell supported on a Au core. J Solid State Electrochem 20, 1753–1764 (2016). https://doi.org/10.1007/s10008-016-3181-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-016-3181-z