
Abstract
Context
Malaria remains a significant global health challenge with emerging resistance to current treatments. Plasmodium falciparum glutathione reductase (PfGR) plays a critical role in the defense mechanisms of malaria parasites against oxidative stress. In this study, we investigate the potential of targeting PfGR with conventional antimalarials and dual drugs combining aminoquinoline derivatives with GR inhibitors, which reveal promising interactions between PfGR and studied drugs. The naphthoquinone Atovaquone demonstrated particularly high affinity and potential dual-mode binding with the enzyme active site and cavity. Furthermore, dual drugs exhibit enhanced binding affinity, suggesting their efficacy in inhibiting PfGR, where the aliphatic ester bond (linker) is essential for effective binding with the enzyme’s active site. Overall, this research provides important insights into the interactions between antimalarial agents and PfGR and encourages further exploration of its role in the mechanisms of action of antimalarials, including dual drugs, to enhance antiparasitic efficacy.
Methods
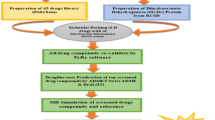
The drugs were tested as PfGR potential inhibitors via molecular docking on AutoDock 4, which was performed based on the preoptimized structures in HF/3-21G-PCM level of theory on ORCA 5. Drug-receptor systems with the most promising binding affinities were then studied with a molecular dynamic’s simulation on AMBER 16. The molecular dynamics simulations were performed with a 100 ns NPT ensemble employing GAFF2 forcefield in the temperature of 310 K, integration time step of 2 fs, and non-bond cutoff distance of 6.0 Å.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Malaria is a condition resulting from the infection by protozoa of the genus Plasmodium (P). These microorganisms can affect birds, reptiles, and mammals, including humans, and there are approximately 200 species of Plasmodium in circulation [1]. Among these species, five are capable of infecting humans and causing the disease: P. falciparum, P. vivax, P. malariae, P. ovale, and, more recently, P. cynomolgi and P. simium. Although the latter two were initially reported in non-human primates, there are records of naturally acquired infection cases in humans [2,3,4].
According to the World Health Organization (WHO), the total number of disease cases reached 249 million in 2023. The majority of cases (82%) and deaths (95%) occurred in the African Region, followed by the Southeast Asia Region (cases 10% and deaths 3%). Recent records indicate that the major contributors to malaria worldwide are the species P. falciparum and P. vivax, with the former responsible for the highest number of fatalities, particularly in the African continent [5]. Regarding Brazil, the Amazon region is indeed considered the endemic epicenter of malaria in the country, accounting for 99% of recorded autochthonous cases. Outside this area, the majority of reported cases originate from endemic states or countries, representing over 80% of cases. In terms of analyses conducted by the Ministry of Health in 2023, there was a decline in malaria notifications between 2010 and 2016. However, in 2017, there was a significant increase of 38.7% compared to the previous year, as highlighted with the blue arrow in Fig. 1 below. In the subsequent years, a reduction in notifications was observed; however, the Ministry of Health raised alarms due to an unexpected increase of approximately 27.8% in 2020, in cases of malaria associated with P. falciparum or mixed P. falciparum/P. vivax malaria (red dots in Fig. 1) [6, 7].

Reported malaria cases in Brazil from 2010 to 2022. The red dots represent the cases of malaria associated with P. falciparum or mixed P. falciparum/P. vivax
Malaria is transmitted to the vertebrate host through the bite of female mosquitoes of the genus Anopheles. The sporozoites from the salivary glands infect the human host through the insect’s bite. In the liver tissues, the sporozoites multiply through asexual reproduction, forming thousands of merozoites. Released into the bloodstream, these invade red blood cells and initiate the asexual erythrocytic phase, causing disease symptoms in the human host after 48 h of infection. Some of the merozoites may develop into male or female gametocytes. When a mosquito bites an infected person, the gametocytes are ingested and mature in its digestive system in around 24 h. Thus, mosquitoes become ready to infect a new host, completing the transmission cycle [8, 9]. During the asexual erythrocytic phase, the parasite digests between 60 and 80% of hemoglobin, transporting it to its digestive vacuole (DV) to process it into an inert and non-toxic material known as hemozoin, which does not affect its survival [10]. In this same phase, the parasite is exposed to oxidative stress generated by the host’s immune system to combat the infection, as well as by the heme group and other decomposition products of hemoglobin [11].
Glutathione (GSH) plays a fundamental role in the defense of malaria parasites against oxidative stress. This tripeptide acts as an antioxidant, aiding the parasite in neutralizing reactive oxygen species both directly and through reactions catalyzed by glutathione peroxidase and glutathione S-transferase [12]. Thus, high levels of reduced GSH are maintained in the erythrocytes of infected hosts (asexual erythrocytic phase) thanks to the action of the enzyme glutathione reductase (GR) [13].
Studies have shown that erythrocytes deficient in GR could fulfill their physiological functions, although with a shorter lifespan. However, they did not serve as host cells for P. falciparum. In fact, the depletion of GSH by GR inhibitors in erythrocytes infected with P. falciparum produces a drastic antiplasmodial effect [12]. Thus, the search for effective inhibitors of PfGR has become a promising strategy for the development of new and effective antimalarial drugs, aiming to overcome current concerns about the disease, given the emergence of strains resistant to first-line treatment (ACT). In addition to the defense of malaria parasites against oxidative stress, studies have highlighted the relationship between GSH and PfGR with the development of resistance to antimalarial drugs [14, 15]. This is supported by the fact that GSH levels increase in mice infected with CQ-resistant strains. Despite CQ treatment not influencing such intracellular GSH levels, the activity of the enzymes GR and GPx (glutathione peroxidase) significantly decreases [12]. Consistently, the depletion of GSH by L-buthionine-(S,R)-sulfoximine (BSO), an inhibitor of GSH synthesis, in resistant strains of P. falciparum, restores sensitivity to CQ and its analogs [15].
The crystal structure of P. falciparum GR (PfGR) was reported in 2003 by Sarma et al. Similar to human GR (hGR), each subunit of PfGR (Fig. 2) has two cysteine residues in the active site (Cys39 and Cys44) that mediate electron transfer [14]. In addition to the active redox pair Cys39/Cys44, the active site includes the cofactor FAD, which is reduced by NADPH after binding to the oxidized enzyme (2NADPH + FAD → 2NADP+ + FADH2 + 2e−). Once reduced, the isoalloxazine ring of FADH− packs against the persulfide, forming a charge transfer complex, reducing the Cys39-Cys44 disulfide. Thus, oxidized glutathione (GSSG) binds to the active site to be reduced (GSH) (GSSG + NADPH+ → NADP+ + 2GSH) and the cysteine redox pair reforms in the disulfide form [13, 16]. Isoalloxazine and 1,4-naphthoquinone derivatives, methylene blue, ajoene, among others, are considered strong competitive inhibitors of both human (hGR) and parasite (PfGR) enzymes, binding to their active sites [17,18,19]. Studies on the mechanisms of inhibitors such as methylene blue (MB) suggest that the enzyme catalyzes the reduction of this compound by FADPH after reduction by NADPH. The resulting reduced species (Leucomethylene blue) is a more efficient auto-oxidant, oxidized by O2. From a cellular pharmacological perspective, each reaction cycle catalyzed by the GR-MB complex leads to the consumption of NADPH and O2, and the production of parasitotoxic reactive oxygen species, predominantly H2O2. Therefore, MB and other redox-active GR inhibitors act as a “subversive substrate” in the enzyme’s active site, oppositely altering its physiological function from reductive/antioxidant to pro-oxidant [17, 20]. Indeed, various studies demonstrated that the most potent and specific effects against malaria parasites in cultures were observed with redox-active agents, acting as “subversive substrates” for both PfGR and hGR from the human host erythrocyte. The area’s researchers stand out that it is essential to continue developing these redox-cyclers since they can affect the thiol equilibrium of parasites. These drugs could be used as new drugs targeting malaria parasites and might replace artemisinin-based combination therapy (ACT) [20].

Crystallographic structure of glutathione reductase from Plasmodium falciparum (subunit; PDB 1ONF) indicating the active site and its key components, along with the cavity located at the enzyme interface
The most pronounced differences between the two enzymes (hGR and PfGR) are found in the cavity region at the dimer interface (Fig. 2), causing changes in the shape and electrostatic properties of this locale [14]. This region defines another binding site for PfGR inhibitors, known as the interface cavity or allosteric site. Unlike the active site, the interface cavity does not confer selectivity regarding ligand binding [21, 22]. Targeting this site might interfere with the dimerization of the enzyme and lead to the conformational change in the enzyme, which might prevent or interrupt the glutathione reduction cycle [14]. This cavity has also been studied for the selective design of antimalarial drugs, which provides advantages such as lowering the likelihood of drug resistance [23]. Inhibitors of PfGR such as Xanthane (6-hydroxy-3-oxo-3H-xanthene-9-propanoic acid) and Menadione (2-methyl-1,4-naphthoquinone) bind to this cavity, potentially causing non-competitive enzyme inhibition [14, 21]. Some studies indicate that MB can also act as a non-competitive inhibitor of GR and PfGR, probably by binding to this enzyme cavity [14, 24].
In response to the challenges posed by parasite resistance and adverse effects, various antimalarial drugs have been developed, targeting different stages of the parasite’s life cycle [25]. These drugs, classified into classes such as aminoquinolines (e.g., chloroquine (CQ), amodiaquine (AQ), primaquine (PQ), mefloquine (MQ)), naphthoquinones (e.g., atovaquone (ATV)), antifolates (e.g., proguanil), and artemisinin derivatives, aim to combat malaria effectively [26]. Several research suggests that 4-aminoquinoline antimalarials and artemisinin derivatives function during the intraerythrocytic stage by binding to the heme group, preventing its sequestration into hemozoin (Hz) [27, 28]. 8-Aminoquinolines like PQ are believed to induce oxidative stress by accumulating H2O2 in the liver, leading to Plasmodium parasite toxicity [29]. Despite these valuable findings, details of this mechanism of action are not well understood to date. The disulfide reductase enzyme PfGR has not been extensively studied as a potential target for these conventional antimalarial drugs. Nonetheless, this enzyme has been identified as a target for 4-aminoquinoline drugs (including CQ) when conjugated with well-known GR inhibitors such as 1,4-naphthoquinones. This conjugation occurs through a metabolically labile ester bond, resulting in the formation of double-headed drugs. These “double-drugs” represent a series of prodrug esters designed and synthesized by Davioud-Charvet et al. as a strategy for overcoming glutathione-dependent 4-aminoquinoline resistance [30, 31]. Among these compounds, the [2-(3-methyl)naphthoquinolyl]alcanoic acids of menadione and plumbagin exhibited the strongest inhibitory activity, where the most active of this last series led to more than 80% inhibition of both hGR and PfGR [30]. Furthermore, computational studies have explored amide-based quinoline hybrids as competitive inhibitors of PfGR. In general, quinoline-containing compounds with 4-amino functionality showed better antimalarial activities than those lacking the 4-amino moiety [32]. Moreover, recent years have seen a surge in computational studies to discover novel Pf reductase inhibitors and delineate a suitable pharmacophore as a starting point for designing drugs targeting this type of parasite’s enzymes [33,34,35].
In the present study, we explore the in silico interaction of quinoline-derived drugs and 1,4-naphthoquinones with the PfGR enzyme through molecular mechanical calculations. Additionally, we investigate double-drugs proposed by Davioud-Charvet et al., combining quinoline-based alcohols with derivatives of 1,4-naphthoquinones, to understand their efficacy in inhibiting PfGR activities. Our discussion, based on binding energy results and graphical analysis of drug-receptor interactions, provides insights valuable for designing potent inhibitors for this essential enzyme in malaria parasites.
Methodology
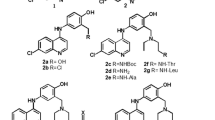
Molecular docking studies were conducted to determine the binding affinity and interactions between candidate inhibitors and the PfGR enzyme (Fig. 2), and some results were better discussed via a molecular dynamics simulation. The aminoquinolines CQ, AQ, PQ, MQ, and the naphthoquinone ATV were selected among commercial drugs available for malaria treatment as ligands for the present study. Additionally, we address the double-drugs strategy proposed by Davioud-Charvet et al. [30]. The double-drugs 1–3 represented in Fig. 3 were also selected as ligands for the present study. The ligands 1c, 3a–c were designed by the authors of the present study. The proposed ligands 3a–c, combine the inhibitor Menadione (MD) directly with quinoline drugs, unlike ligands 1 and 2, which have an ester bridge with a variable number of carbon atoms in the aliphatic part (n) connecting both residues as depicted in Fig. 3.

Molecular structures of conventional antimalarials and double-drugs combining quinoline-based with 1,4-naphthoquinones studied in the present work
Preparation of ligands and receptor
The structures of conventional antimalarial drugs (CQ, AQ, PQ, MQ, and ATV; Fig. 3) were obtained from the CCDC repository [36,37,38,39,40] via crystallographic approaches and the proposed hybrid species were built using Avogadro visualization software [41]. All drugs are represented in Fig. 3 and were optimized and characterized as minima on the potential energy surface in aqueous phase at the Hartree–Fock level, using the 3-21G basis set in the ORCA 5.0.3 program [42]. The optimization calculation was performed in order to keep consistency between the generated structures and the ones obtained from experiments. The solvent effect was accounted for using the CPCM method [43]. The choice of a more basic method is justified by the subsequent treatment by molecular mechanics methods since all structural properties are parameterized by the force field, and the initial structures only provide a reference for the program without requiring an advanced description of the electronic part.
The protein receptor PfGR enzyme was obtained from the Protein Data Bank (PDB) under the code 1ONF [14] and edited using Discovery Studio software [44]. The structure had the crystallographic water molecules removed, and the residues were renamed based on the pdf4amber software package available in the AMBER [45, 46] suite. Concerning the enzyme’s cofactors, FADH2 was kept near the active site of the biomolecule, which was reconstructed based on the incomplete structure present in the original file.
Molecular docking
Molecular docking simulations were conducted for all ligands using the software package Autodock 4.2.6. [47]. Two regions were selected for structural testing, the enzyme’s active site (defined here as site 1) and the intersubunit cavity (defined as site 2), as depicted in Fig. 2. Both were determined at specific locations by fixing a rectangular box, measuring 60 Å-70 Å-60 Å and centered at x:73.371; y:67.404; z:80.181 for site 1, and measuring 50 Å-100 Å-60 Å and centered at x:62.671; y:38.441; z:88.822 for site 2, with a spacing of 0.4 Å. Lamarckian algorithms were employed to obtain 20 target-ligand interaction poses in a population of 250 structures. Images of the poses and vdW surfaces were generated using Discovery Studio Visualizer 2019 software [44] and Chimera 1.17.3 [48].
Molecular dynamics
Force-field parametrization
The parameters for the PfGR enzyme atoms were extracted from the FF14SB force field, while those for the ligands (Fig. 3) were derived from GAFF2 [49, 50]. A total of 22,250 explicit water molecules were loaded as the solvent within a periodic boundary condition, structured as a truncated octahedron box. The solvent was represented using the 3-site rigid water TIP3P model [51], with a box radius of 15.0 angstroms. Six chloride ions were included to neutralize the protein’s net charge. Overall, the simulations involved approximately 74,000 atoms, varying based on the size of the ligands.
Simulation protocol
The simulation protocol comprised two successive minimization steps: the initial step involved steepest descent cycles (1000) followed by cycles of conjugated gradients (1500). During the first minimization step, a restraint force constant of 500 kcal mol-1 Å-2 was applied to the solute, while in the second step, no restraint was imposed, allowing the entire system to undergo unconstrained minimization. Subsequently, six intercalated heating and equilibrium steps were conducted, each involving a temperature rise of 50 K, except for the final step, which increased by 60 K, culminating in a temperature of 310 K. These steps were executed under constant volume periodic boundaries (NVT) over 2800 cycles (2 fs in time), with frames of equilibrium (2 fs) implemented under constant pressure periodic boundary conditions (NTP) at a mean pressure of 1 bar. The production phase extended over 100 ns, comprising frames with a temporal interval of 2 fs between each frame and a cutoff distance of 6.0 Å for non-bonded interactions. The simulation was conducted under constant pressure and temperature (NPT) conditions, with the temperature maintained at approximately 310 K through the implementation of a Langevin thermostat and the pressure regulated at 1 bar using the Berendsen barostat [52, 53] in order to simulate physiological condition. The SHAKE [54] algorithm was activated to restrict hydrogen stretching during the simulation. All molecular mechanics simulations were performed in Amber 16 [45, 46].
Binding free energy calculations
The ligand-PfGR binding free energies (\({\Delta G}_{bind}\)) from docking were predicted to rank ligand interaction poses. AutoDock semi-empirical force field works with pairwise components such as ligand (L) and protein (P) in bound (b) or unbound (unb) state, as shown in Eq. (1), to properly estimate this parameter. Each term contains four contributions that are followed by a weighting constant (W) optimized based on a set of experimentally determined constants. In total, it is accounted contributions from the well-potential in the first term, hydrogen bond contributions, and the function E(t), which control the angle t for ideal H-bonding geometry in the second, coulombic electrostatic potential in the third, and a final parameter based on the solvation-free energy contribution as represented in Eq. (2). This total \({\Delta G}_{bind}\) parameter is the determining point in the Lamarckian algorithm as it will decide which structures are “better interacting” and will be considered in the next set of poses until the best 20 are localized.
Where
The inhibition constant (Ki) is calculated as \({K}_{i}={e}^{\frac{\Delta G}{RT}}\) where ∆G is the free energy of binding, R is the gas constant (1.987 cal mol−1 K−1), and T is the temperature (298.15 K). The smaller the value of Ki, the lower the probability of complex dissociation and, therefore, the higher the inhibition.
Regarding the molecular dynamics production, trajectories were analyzed by using the cpptraj tools [55] and were employed to also estimate the ligand-receptor binding energies through the Generalized Born/Surface Area model MM-GBSA, accessible through the MM-PBSA.py script [56]. These energies for binding of the ligand (lig) to the enzyme receptor (rec) to form the complex (lig-rec) are basically computed by solving the following Eq. (3). This \({\Delta G}_{bind}\) can be decomposed into individual contributions and represented as in Eq. (4):
In Eq. (4), \({\Delta E}_{MM}\) refer to the gas phase molecular mechanical energy, \({\Delta G}_{sol}\) is the solvation free energy calculated implicitly, and \(T\Delta S\) denotes the entropy of the system upon ligand binding. The gas-phase molecular mechanics energy \(({\Delta E}_{MM})\) includes variations in internal energies (bond, angle, and dihedral energies), as well as electrostatic and Van Der Waals energies. \({\Delta G}_{sol}\) is defined as the sum of the polar and the nonpolar contribution. The polar contribution is calculated using the GB model, while the latter is commonly estimated using the solvent-accessible surface area (SASA). The entropy change is determined by normal-mode analysis, decomposed in contributions related to its translational, rotational, and vibrational [57].
Results and discussion
Selected bond distances and angles for ligand structures CQ, AQ, PQ, MQ, and ATV were summarized in Tables S1-S5, wherein calculated and experimental data were compared. In general, the geometric parameters of optimized structures at the HF level were in agreement with the solid structures available on the CCDC Cambridge website [36,37,38,39,40]. Bond lengths were overestimated in aqueous solution, with a maximum error of 5.2% and a maximum deviation of 0.09 Å in relation to the solid state. The error falls within the margin of the HF method, partly due to the solvent effect that tends to increase bond lengths in aqueous solution. Angular parameters also show satisfactory agreement with experimental values, with a maximum error of 4.1%.
Evaluation of conventional antimalarial drugs as PfGR inhibitors
Initially, molecular docking studies were conducted with the drugs CQ, AQ, PQ, MQ, and ATV, compounds of pharmacological significance, particularly in the treatment of malaria. CQ and AQ are weak diprotic bases; therefore, at physiological pH (∼7.2), they can exist in non-protonated, monoprotonated, and diprotonated forms [58]. Thus, the effect of protonation on these two drugs was investigated.
Interaction studies in the active site (site 1)
The molecular docking data at the enzyme’s active site are reported in Table 1. The binding energies and estimated constants of Table 1 indicate a higher affinity of the studied antimalarials when compared to reference inhibitors MD and MB. All the studied drugs exhibited a binding energy score (\({\Delta G}_{bind}\)) lower than -6.0 kcal mol−1 (except for CQ in the neutral form). Despite the neutral CQ showing the lowest scoring of the (\({\Delta G}_{bind}\)= − 5.89 kcal mol−1), among the series of antimalarials studied, this value is quite close to reference inhibitors MD (− 5.53 kcal mol−1) and MB (− 6.08 kcal mol−1).
In its most stable state, neutral chloroquine (CQ) (in orange; Fig. 4a) adopts an extended conformation. The quinoline ring is perpendicular to the isoalloxazine ring, engaging in a π-π T-shaped interaction (4.6 Å) and an N–H···Ph hydrogen bond (2.5 Å) with the drug’s pyridine π electron cloud (Fig. 4a). H-bonds with Val383 assist in positioning the quinoline ring toward FADH2 (Fig. 4a). Moreover, the diethyl-pentane-amine group approaches Cys44, essential for the enzyme’s electron transfer intermediate, through carbon-hydrogen bonds and hydrophobic interactions (Fig. 4a). As expected, increasing protonation in CQ leads to more H-bond formation, reflected in the elevated score (Table 1, Figure S1). In the monoprotonated (CQH) and diprotonated (CQH2) forms, a more closed conformation is observed, especially in the CQH form with a higher RMSD value of 3.23 Å compared to the neutral (1.90 Å) and diprotonated (1.94 Å) forms. In the diprotonated form (green; Fig. 4a), the quinoline ring points toward the enzyme surface, interacting with residues in the α-helix. Conversely, in the CQH form (navy blue), the structure inverts, directing the quinoline ring toward the interface and interacting with residues in the β-sheets (Fig. 4a).

Binding mode of CQ, AQ, and ATV after docking at the active site of PfGR. 3D structures of the drug-receptor complexes are presented highlighting the drug conformation and interactions with the main residues in direct contact with the ligand. The Van Der Waals surface of the enzyme is also shown
Like CQH2, AQ’s quinoline ring approaches the FADH2 while its diethylamino-phenol group moves toward the interface region (Fig. 4b). Only the monoprotonated form interacts with FADH2, establishing weak π-π stacked interactions between the flavin and quinoline rings and a π-alkyl interaction involving the drug’s chloride (Figure S2).
Amodiaquine in the diprotonated form (AQH2) scores slightly better than the neutral (AQ) and monoprotonated (AQH) forms, displaying the lowest binding energy (-7.39 kcal mol−1). This is attributed to the formation of strong H-bonds between the diethylamino-phenol group with residues Asp458, Glu459, and Gln462 (⁓2.0 Å) of the first α-helix in this interface (Fig. 4b; Table 1). Moreover, the electrostatic contribution increased, resulting from interactions of the diethylaminomethyl-phenol group with residues Lys48 and Glu459 in the interface (Table 1; Fig. 4b) and a better fit into the cavity of this region (Fig. 4b). Protonation has a more pronounced effect on the lead drug CQ than its derivative AQ, causing significant conformational changes and larger scoring alterations (Table 1). The 8-aminoquinoline PQ adopts a conformation similar to CQ and AQ. The methoxy group of the quinoline ring interacts weakly with FADH2 through a π-alkyl bond (4.1 Å). The alkyl portion with the amine function heads towards the protein interface, forming H-bonds and hydrophobic interactions with the same residues involved in interactions with CQ and AQ (Table 1).
The MQ has two stereogenic centers, but its erythro racemic mixture ((11S,12R) and (11R,12S)) is used clinically [64]. The drug’s stereochemistry was considered for the docking study. Both MQ enantiomers exhibited similar interaction energy values at the PfGR active site. However, (R,S)-MQ showed a slightly better score, boasting an interaction energy of -6.59 kcal mol−1 and an inhibition constant of 14.74 µM (Table 1). In the lowest energy orientation, the piperidine approaches FADH2, forming a robust H-bond (1.89 Å) between the oxygen of FADH2 and the nitrogen of the piperidine ring of MQ. Simultaneously, the quinoline portion with two –CF3 groups addresses the enzyme interface, showcasing notable halogen bonding interactions F∙∙O (2.4 Å) and F∙∙N (2.9 Å), especially with Pro381(Figure S3).
The hydroxy-1,4-naphthoquinone ATV, in docking analysis, exhibits a \({\Delta G}_{bind}\) value of − 8.10 kcal mol−1 (best scoring), showcasing the highest affinity for the enzyme’s active site. The estimated inhibition constant value (1.15 µM) is in agreement with reported values for 1,4-naphthoquinone in PfGR inhibition (2.2 µM) and hGR (1.3 µM) [18]. Similar to neutral CQ, ATV’s naphthoquinone ring is oriented inward, perpendicular to the isoalloxazine ring (Fig. 4c). In fact, the same interactions are found: an H-bond N–H···Ph and a π-π (T-shaped) interaction of 4.63 Å between the isoalloxazine and quinoline rings. However, an extra weak amino-hydroxide H-bond (N–H∙∙OH; 3.08 Å) stabilizes the ligand-receptor arrangement (see Fig. 4c). More H-bonds with Val383 (Table 1) are formed in this system, aiding in positioning the NQ ring toward FADH2. Moreover, the chlorophenyl-cyclohexyl group adopts a conformation against the plane of the NQ ring, weakly interacting with initial residues of the α-helix, including Cys44. This arrangement strengthens interactions at site 1, positioning the ligand close to FADH2 and the active redox pair Cys39/Cys44, with ATV showing stronger interactions than CQ.
In the docking analysis for the MB inhibitor (Fig. 5), the best-scoring orientations are closely similar to the positioning of the quinoline rings of CQ and the 1,4-NQ of ATV in the FADH2 binding site (Fig. 4a and 4c). Similar aromatic ring interactions (π-π, T-shaped) with FADH2 and Val383 from the β-sheet are observed (Figs. 4a, c, and 5). In contrast, MD positions itself farther from FADH2 (⁓4.9 Å) and weakly interacts with Val383, resulting in a lower score than MB (− 5.65 kcal mol−1). The Michaelis constants (Km) for PfGR inhibition, obtained by following NADPH oxidation (42.2 µM for MB and 82.2 µM for Menadione), are in agreement with those estimated in the docking study (35.5 µM and 88.6 µM, respectively; Table 1) [63].

Methylene blue binding modes with the active site of PfGR. 3D structures of the drug-receptor complexes are presented, showing the drug conformation and interactions with the main residues in direct contact with the ligand. The Van Der Waals surface of the enzyme is also presented
According to the vdW surface of the enzyme in Figs. 4a and 4c, it is noted that CQ and ATV dock similarly to the active site of the enzyme. It is observed that neither of the two ligands penetrate the cavity above FADH2 (Figs. 4a and 4c). In the case of CQ, the presence of a substituent bulkier larger than chloride could fit better into this cavity, potentially providing extra stability to the drug-receptor complex. Analogously for ATV, with a substituent in positions 7 or 8 of the NQ ring. This cavity is partially filled by the dimethylamino substituent in the reference inhibitor MB, according to the second-best binding pose in this active site (pose 2; Fig. 5).
It is worth highlighting that 1,4-naphthoquinones such as ATV are redox-active compounds and can act as acceptors of electrons from different flavoproteins like glutathione-disulfide reductase. The reduction of these compounds by these latter enzymes results in the formation of semiquinone radicals or quinone dianion. These species lead to the generation of superoxide and peroxide through oxygen reduction and, ultimately, the regeneration of naphthoquinone [20]. Nevertheless, previous studies have emphasized that the reduction potential of ATV is low for efficient two-electron reduction under intracellular conditions (⁓ − 0.26 V vs NADP) [19]. In contrast, the two-electron reduction potential for methylene blue is estimated at ⁓ − 0.01 V and ⁓ − 0.25 V for MD at pH 7.
Given that the activity of structurally diverse antimalarial agents against P. falciparum may be related to their unique redox properties [18,19,20, 65, 66], we investigated their redox character. The standard two-electron reduction potential (Eo) relative to the NADPH/NADP+ redox couple at pH = 7 was calculated for the antimalarial conventional drugs (Fig. 3) together with the inhibitors MD and MB. The Eo was calculated in aqueous solution at level B3LYP/6–31 + G(d) [67, 68] (see more details in Supplementary Information).
Although the estimated Eo values of ATV, MD, and MB were underestimated (Fig. 6), the reduction trend is in agreement with the reported data. Overall, the reduction potential increased in the order PQ < AQ < AQH < MQ < CQH2 < AQH2 < MD < ATV < CQH < CQ < MB. MB exhibits the highest reduction potential in the studied series (-0.34 V), followed by CQ (− 0.63 V) and ATV (− 0.76 V), whose Eo value is pretty close to that estimated for the inhibitor MD (− 0.81 V), as shown experimentally [19]. MQ exhibited a redox stability similar to CQH2, while PQ was the most stable drug with the lowest Eo in the series (Fig. 6).

Predicted reduction potentials of antimalarial drugs vs NADPH/NADP+ redox couple
The chloroquine reduction potential decreases significantly for the diprotonated form (CQH2) (− 1.38 V), where the electron density is more concentrated over the quinoline ring for this full protonation form, as evidenced by the molecular electrostatic potential maps (MEP) of Figure S4. A similar reduction is observed for AQH2, with a slightly higher Eo (− 1.13 V). For both reduced species, the greatest charge variation is observed in the 4-amino pyridine group, confirming its active participation in the reduction. The N–C2 bond length increases while C2–C3 decreases, showing the loss of aromaticity and the formation of a structure with a localized double bond on the pyridine ring as in the quinolidine anion structure. Distortions in the planar quinoline ring geometry are evident in these reduced structures (Figures S4 and S5).
CQ and CQH exhibited a greater tendency to reduction (Eo⁓ 0.64 V) than their analogs AQ and AQH (Eo < − 2.0 V). After CQ and CQH reduction, the C7‒Cl bond is broken, observing a high charge density on the quinoline ring’s C7 and the leaving chloride, where the minimum MEP is found (Figures S4). In contrast, chlorine remains attached to the quinoline ring after the reduction of AQ and AQH, with no significant distortion of planarity observed. The electron density is distributed mainly on the p-hydroxyanilino and pyridine aromatic rings (Figure S5), disfavoring the reduction in relation to CQ and CQH2.
Concerning ATV and MD, the formation of the quinone dianion occurs after a two-electron reduction, leading to structural modifications over the 1,4-dione ring. Similar structural changes over the quinoline ring are observed after the reduction of MQ and PQ (Figure S6).
Such electrochemical results suggest that ATV and CQ may not be as efficient “subversive substrates” as MB; however, they exhibit slightly higher Eo values to MD, indicating similar redox features to this inhibitor, which is considered a moderate redox-cycler drug [18]. These similar redox characteristics of ATV and CQ with the inhibitor MD, along with their observed docking mode in the enzyme’s active site, are interesting. The ligand-receptor arrangement promotes contacts necessary to position the ligand in a more lipophilic region near FADH2 and the active redox pair Cys39/Cys44 (Fig. 4a,c).
This docking arrangement could favor drug reduction since studies have evidenced that flavin (FADH2), rather than the thiols, mediates the reduction [19]. However, in the case of CQ, interactions with FADH2 and Cys44 are weaker, and protonation disfavors its reduction, impacting its efficacy as a subversive substrate.
Interaction studies in the intersubunit cavity (site 2)
In addition to its active site, the interface region’s cavity is considered another binding site of the PfGR enzyme. The interaction of the same ligands in Fig. 3 with the intersubunit cavity (site 2) was analyzed, and the results are presented in Table 2. The docking evaluation results again reveal ATV to have the highest affinity for this site 2, with a free binding energy of -9.28 kcal mol−1 and an inhibition constant of 156.51 nM (Table 2). It is noteworthy that all the ligands analyzed increased their interaction energy values compared to site 1 and outperformed the scores for the inhibitors MD, Xanthane, and MB, except for PQ (Table 2). Nevertheless, a consistent trend in the stability of the drug-PfGR complex at the active site is observed in the cavity (ATV > AQ > MQ⁓CQ > PQ). These results suggest a higher affinity of antimalarial drugs for the cavity when compared to the active site.
IC50 data for PfGR inhibition by conventional antimalarial drugs are unavailable in the literature. Davioud-Charve et al. explored this property for ATV and analogs, reporting capabilities below 25 μM [65]. However, precise IC50 values were hindered by compound precipitation in solution at doses exceeding 25 µM. To compensate for this absence of data, in vitro antiplasmodial activities against CQ-susceptible and resistant strains were included in Tables 1 and 2. These results demonstrate a notable correlation with docking interaction energies, where drugs with stronger antiplasmodial activity exhibit lower binding energy (better score), particularly in their interaction with the enzyme cavity (site 2). The correlation in this second binding site reveals a correlation coefficient, R2 ⁓0.75 (Figure S7), observed in both CQ-sensitive and resistant strains.
All antimalarial drugs, including the MD inhibitor, dock within the same region of the cavity (Fig. 7). The docking takes place in a more hydrophilic between the final residues of the enzyme’s largest α-helix from the FADH2 binding domain, linking the cavity with the active site (in blue, Fig. 7), and the sequence of parallel β-sheets in the interface (in red, Fig. 7). ATV and AQ have the ability to reach the innermost part of the enzyme by interacting with residues in the NADPH-binding domain region (in gray, Fig. 7) and the connecting loop between the last two β-sheets of the interface. Conversely, the inhibitor Xantane and MB are positioned at the monomer interface, proximate to the last α-helix and the β-sheet of this region (Figure S8). A similar binding position of inhibitor Xantane is found at the hGR dimer crystal structure [14].

Complexes PfGR-antimalarials in their most favorable arrangement in the enzyme cavity
In general, the complexation of the drugs with the cavity is generally stabilized by a greater number of H-bonds and electrostatic interactions due to the hydrophilic character of the region (Table 2). Concerning chloroquine, the monoprotonated form CQH demonstrates the most favorable interaction with the cavity (\({\Delta G}_{bind}\)= -6.78 kcal mol−1; Table 2). In contrast to the active site, CQH positions its quinoline ring towards the enzyme surface, engaging in H-bonds with residues from the β-sheets of the interface (Asn456) and the largest α-helix (Ser55) (Fig. 7). Π-Sigma interactions with Leu455 and π-cation interactions with His387 contribute to the quinoline ring stabilization. Although the aliphatic chain has minimal interaction with the cavity, charge attractions occur between Glu432 and the protonated nitrogen of the tertiary amine within the cavity (Fig. 8a). Notably, the monoprotonated form establishes the highest number of contacts with the enzyme (Table 2), predominantly through its well-fitted quinoline ring on the cavity’s surface (Fig. 8a), enhancing the stability of the PfGR-CQH complex.

Binding mode of CQH (a), AQH2 (b), MQ (c), and ATV (d) after docking into the PfGR cavity. 3D structures of the drug-receptor complexes are presented, showing the drug conformation and interactions with the main residues in direct contact with the ligand. The Van Der Waals surface of the enzyme is also presented
In the protonated forms, the amodiaquine quinoline ring deepens into the cavity’s inner part with the diethylamino-phenol group towards the interface’s last two β-sheets (see Figs. 7 and 8b). In contrast, the neutral form of amodiaquine assumes a different orientation, placing its quinoline ring on the enzyme’s surface. Simultaneously, the diethylamino-phenol group shifts towards the cavity’s interior, establishing H-bond interactions with Lys228 and Asp225 (see Table 2 and Figure S9).
The AQH2 form maintains its extended conformation upon interacting with the cavity, exhibiting minimal structural deformations. Its RMSD value is significantly smaller at 1.78 Å compared to AQH (4.70 Å) and AQ (4.48 Å). Both protonated structures’ OH and secondary NH groups act as H-bond acceptors with residues Ser55 and Glu432 (⁓2.1 Å). Moreover, the additional proton from the quinoline ring in AQH2 engages in an H-bond with Asn62 (2.3 Å), a terminal residue of the α-helix (Fig. 8b).
Protonation considerably increases electrostatic energy, resulting in a 0.81 kcal mol−1 increase between the neutral and di-protonated forms. Notably, protonated states exhibit a higher number of charge attractions between the diethylamino-phenol group and Asp58 and Glu432 (Table 2 and Fig. 8b). These interactions significantly contribute to the better binding score of AQH2 (\({\Delta G}_{bind}\)= − 8.61) compared to the neutral form (\({\Delta G}_{bind}\)= -7.83). The effect of protonation on chloroquine and amodiaquine becomes more pronounced in this secondary binding site, characterized by a more lipophilic nature.
For this second site, the (S,R) enantiomer of MQ exhibited high score than (R,S) counterpart. The conformation adopted by (S,R)-MQ enhances contact and forms numerous hydrogen bonds with cavity residues, significantly contributing to the free interaction energy. Similar to CQH and neutral AQ, the quinoline ring of (S,R)-MQ, with its two –CF3 groups, is oriented towards the surface (Fig. 7). Here, fluorine acts as a hydrogen bond acceptor with Asn456 of the interface and Ser55 of the α-helix (Fig. 8c). Additionally, the –CF3 substituents engage in halogen interactions. The 2-piperidyl-methanol group also contributes to stabilizing the conformation through hydrogen bonds with Ser55 and Glu432 (Fig. 8c). However, the ligand structure of (S,R)-MQ does not conform to the cavity shape or align with the site compared to CQH and ATV (see vdW surface in Fig. 8c).
Docking results highlight ATV’s tighter binding to the enzyme’s cavity compared to the other studied drugs. The 1,4-NQ ring of ATV penetrates the cavity, reaching its innermost part (Fig. 7). The carbonyl oxygen serves as a hydrogen bond acceptor with internal residues Arg196 and Asn229, while the hydroxyl is directed towards the major α-helix, forming a robust hydrogen bond with Asp58 (OH∙∙∙O = CO; 2.0 Å) (Fig. 8d). Notably, residues such as Asp58, Asn62, His65, and Arg196 in PfGR, as identified by Sarma et al., contribute to altering the electrostatic properties of the cavity compared to the analogous human enzyme (hGR) [14]. Moreover, the NQ ring is stabilized by electrostatic interactions (π-ion), particularly with residues Asp58 (4.87 Å) and Glu432 (2.97 Å) (Fig. 8d), influencing the drug’s positioning within the cavity. The same interactions are observed for the naphthoquinone ring of the reference drug MD; However, the charge attraction is weaker with Glu432 (3.67 Å) (Figure S10), and the lack of the extra polar group hydroxide as in ATV disfavor significantly the H-bond formation inside the cavity. Furthermore, the ATV’s chlorophenyl-cyclohexyl group stretches across site 2, occupying a substantial cavity region and contributing to its heightened stability (see vdW surface Fig. 8d).
ATV stood out as the drug with the highest score for both investigated binding sites, showcasing as the best candidate to inhibit PfGR between the series of conventional antimalarial drugs studied. The potent antimalarial activity of ATV against both sensitive and resistant parasite strains might involve the inhibition of PfGR, complementing its established action mechanism, specifically, inhibiting the mitochondrial bc1 complex of P. falciparum and disrupting the mitochondrial electron transport chain [69]. This inhibition is likely attributed to the drug’s binding in both the active site and the cavity within the interface region, exhibiting a dual docking mode.
Antimalarial dual drugs based on inhibitors of PfGR
Analysis via molecular docking simulations
Table 3 presents the energetic data obtained from molecular docking simulations, along with experimental ED50 and inhibition percentage data for dual drug compounds [30]. Compounds 1a, 1b, and 1c are all naphthoquinones that share the same number of carbons in the 3-ester linker, comprising different O–R substituents. While in 1a, naphthoquinone position 3 is linked to a pentoic acid, 1b and 1c are occupied by chloroquine and amodiaquine, respectively. An amodiaquine-like substituent is employed in compounds 1(d–f), varying the length of the linking ester between the naphthoquinone motif and the aminoquinoline ring. The influence of an –OH group (Plumbagin-based compounds) in the naphthoquinone skeleton was also evaluated through compounds 2a and 2b, which are direct analogs of 1e and 1f, respectively. Additionally, the direct naphthoquinone-aminoquinoline dual drugs are under the names of 1(a–c), which comprise (3a) chloroquine, (3b) amodiaquine, and (3c) the amodiaquine-like compound. These dual drugs showed high stability toward chemical hydrolysis in aqueous solutions and under physiologic conditions [30].
The best molecular docking binding energy achieved for site 1 comprises compound 1d (n = 2 / − 9.77 kcal mol−1), with the shortest alkane chain among the ester-linkers. In the same group, the binding energy reduced with the increase of carbons in the ester bridge: compound 1e (n = 4 / − 9.09 kcal mol−1) and 1f (n = 5 / − 7.93 kcal mol−1).
On the other hand, 1e and 1f showed a slightly better score than 1d, whose binding energy did not vary significantly (⁓ 9.60 kcal mol−1) for site 2. A similar tendency is observed experimentally, where both P. falciparum inhibition and ED50 increase with the ester length; nonetheless, such an increase of n = 4 for n = 5 produces slight biological effects (Table 3). This might be explained by the differences in the permeability in the cellular membrane as hydrophobicity may increase with the length of the chain, allowing more of the compound to reach the active target, thus presenting higher toxicity to the parasite cell.
Furthermore, among the compounds with five carbons in the alkane chain, 1b [the double drug containing chloroquine] presented an ED50 of 0.107 µM. However, the docking interaction energy was only − 4.43 kcal mol−1 in the protein’s active site. In this instance, the discordance suggests that the conjugation strategy involving an NQ alkanoic acid might instigate a synergistic effect, leading to the accumulation of the ester in the parasite’s food vacuole. This accumulation amplifies the interaction with heme groups, thus impeding the formation of inert hemozoin crystals rather than obstructing the biological function of PfGR. 1b presented − 6.34 kcal mol−1 interaction energy in the allosteric site, which is in the mean of the other compounds in the group.
In the plumbagin compounds (2a and 2b), both performed well in the molecular docking tests interacting with site 2 with energies of − 8.65 and − 8.32 kcal mol−1, respectively. 2a was the most favored in relation to the active site (− 9.00 kcal mol−1) what might infer the lower ED50 concentration in relation to 2b. Again, there is a direct connection between the reported values of ED50 and the calculated Ki for the compounds.
Compounds 1f and 3c differ only by the linking chain that connects the naphthoquinone group with the quinolinic condensed rings. While the first is connected through an ester functional, the second is bound via a simple alkane chain. 1f demonstrated activity in the 0.0023 µM of ED50, which is concise to the 0.096 µM inhibition constant based on the − 9.58 kcal mol−1 interaction with the allosteric site 2. Thus, for compound 1f the simulations agreed with experimental in vitro and in vivo tests [30] that demonstrated 1f as the most promising inhibitor among the ones studied.
Besides, 3d displayed an interaction energy with site 2 of − 9.28 kcal mol−1 and is indicated, thus, as a possible candidate for biological tests to investigate its antiparasitic properties further. Compared to the currently employed inhibitor MD, all studied compounds (but 1b in site 1) displayed lower binding energy, indicating they might be more effective PfGR inhibitors. This highlights the potential of combining drugs to design better analogs that positively impact some treatments’ efficiency.
Molecular dynamics simulations of the dual drugs 1f and 3c were performed for the best pose in docking in order to explore the binding modes at thermodynamic conditions and validate the docking results.
Analysis via molecular dynamics simulations
The molecular dynamics simulations allowed a more detailed analysis of compounds 1f and 3c, mainly about the reference MD. The estimated average binding free energies were also reported in Table 3 in parenthesis, where data point out the dual drugs with a significantly greater binding affinity than inhibitor MD. The thermodynamic analysis in the realm of molecular docking had predicted a better affinity of 3c than 1f for site 1. This binding energy was corrected by the molecular dynamics GBSA profile which predicted an interaction energetics of − 57.42 kcal mol−1, which is almost twice as favored as 3c (− 32.74 kcal mol−1) (Table 3). However, regarding site 2, this interaction energy difference lowers to a 9.2 kcal mol−1 (1f: − 54.82 kcal mol−1; 3c: -45.62 kcal mol−1), presenting a more modest variation, which is closer to the data obtained from molecular docking analyses.
Along the molecular simulation trajectory, some insightful conformations were observable. At first, 1f interaction on site 1 was predicted to occur mainly via the residue Val383 (as observed for the conventional antimalarials and the inhibitor MB), which acts as an arm that appropriately allocates the ligand in the binding position. That supposition was confirmed by the molecular dynamics, which described the ester oxygen interacting uninterruptedly with the amine group in the protein residue. In the first 3 ns of the simulation, the conformation obtained in the molecular docking for this binding mode rotated in the esters’ C–O bond, resulting in a new conformation. Figure 9a indicates the interaction poses on both molecular docking and dynamics. Whereas in the docking-obtained structure, the naphthoquinone part of the ligand went towards the NADPH-binding domain, and the amodiaquine part was found near the FADH2 binding domain, in the molecular dynamics, after 3 ns, the rotation of the ester group lead both drug skeleton to a more hydrophobic region.

Binding mode of 1f (a) and 3c (b) in the molecular docking and molecular dynamics simulation in the enzyme active site (site 1). The Van Der Waals surface of the enzyme is also presented
Differently, ligand 3c keeps the interaction profile observed in the molecular docking simulation on site 1, and the only observable contrast in the molecular dynamics is a movement away from the FADH2. This movement allows the ligand to move to a more hydrophilic a part of the protein surface, which is stabilized by an H-bonds between the carboxylate in NQ and the residue GLN462. Figure 9b demonstrates the binding mode described above. That difference in binding mode between 1f and 3c may infer the importance of the ester group, which interacts directly with FADH2 in the case of 1f and keeps the drug positioned optimally in the active site of the PfGR. The lack of a hydrogen acceptor group in the alkane chain of 3c makes it translate to a less effective area, thus reducing the GBSA interaction energy (1f: − 57.42 kcal mol−1; 3c: − 32.74 kcal mol−1; Table 3).
On the other hand, 1f interacted with the cavity of the enzyme (site 2) mainly via the interface area. Here, the linking chain was merely a better tool to optimally position both terminal drug structures. The long chain allows both terminal drugs to interact better with the interface region and fit more suitably with the enzyme shape (Fig. 10a,b). The idea for testing the interaction in this cavity is to study the viability of a protein dimerization blockage once this is the area that connects both monomers in the dimer unit. Additionally, as the cavity is more hydrophilic than the active site, the increase in the number of polar groups (such as the ester) may favor the binding energy, as is the case of 1f over 3c (Fig. 10b,c). This energetic behavior on site 2 is also shown in Table 3 (1f: − 54.85 kcal mol−1; 3c − 45.62 kcal mol−1). Besides, 3c interaction geometry did not change in relation to the molecular docking simulation. In the case of site 2, the shorter linking chain of 3c was not affected significantly in the interaction mode, and a longer linker (1f) only led to a slightly better proximity with the protein residues.

Binding mode of 1f and 3c in the molecular docking and molecular dynamics simulation in the enzyme cavity (site 2) (a). 3D structures of the 1f-PfGR (b) and 3c-PfGR (c) complexes are presented, showing the drug conformation and interactions with the main residues in direct contact with the ligand. The Van Der Waals surface of the enzyme is also presented
Some deviation analysis through an RMSD approach indicates that, in general, the protein residues have a reduction in structural freedom when interacting with the drugs compared to a simulation comprising only the protein and its cofactor. 1f interaction in the active site demonstrates an RMSD mean of 3.84 Å, whereas 3c in the same location accounted for a mean of 4.80 Å. This difference infers the binding effectiveness of each compound in PfGR and may indicate the importance of a more flexible and polar linking chain (1f: 5 carbons-ester long; 3c: 2 carbons-alkane long). Similarly, a RMSD analysis in the MD and reference resulted in 4.61 Å and 5.95 Å, respectively. Regarding the interaction influence over the FADH2 RMSD, 1f causes both the major deviations and for longer than 3c (Figure S11). This might be caused by the shorter proximity between 1f and the cofactor, once 3c, as discussed earlier, tends to move away from the FADH2 domain.
Furthermore, when interacting in the cavity (site 2), a contrary behavior was observed: 1f reduced the protein RMSD by 4.29 Å compared to a reduction of 3.61 Å achieved by 3c. MD had a slightly better performance than the one for the active site and displayed a protein RMSD of 4.45 Å. All RMSD graphs are displayed in Figure S11 in the supplementary information. Similarly, a solvent-accessible surface area analysis (SASA) demonstrated a mean area available to the solvent of 25,767.91 Å2 on the protein during the reference simulation. This average was lower for the compound interacting on site 2 (1f: 23,859.96 Å2; 3c: 23,097.26 Å2) than for site 1 (1f: 24,075.94 Å2; 3c: 23,954.61 Å2). However, MD behaved differently, and the SASA for site 1 (24,210.24 Å2) was lower than the one achieved interacting with site 2 (24,435.59 Å2). All SASA graphs are displayed in Figure S12 in the supplementary information.
A root mean square of atomic fluctuation (RMSF) was also evaluated, and the analysis of the residues along both sites of interest demonstrates the binding effect of the drugs based on the fluctuation of atoms in the protein. All drugs can decrease RMSF in the protein in comparison to the free protein simulations. The highest decrease was, in general, achieved by both compounds 1f and 3c, which was better than MD in most studied frames. This very same pattern appeared on site 2 RMSF analysis. Although dual-drugs 1f and 3c performed quite similarly in both sites, 1f binding was slightly more effective in the active site than 3c. On the other hand, 3c performed slightly better on site 2, according to the RMSF results. All RMSF graphs are displayed in Figures S13-14 in the supplementary information.
In summary, a significative change was not observed in the interacting pose, which demonstrates the affinity of those simulated dual drugs and PfGR. Figures S15-16 in the supplemental material illustrate that even though there is a fluctuation in both CQ condensed rings and MD motifs, the overall binding conformation is kept, and the drugs do not rotate inside the binding sites.
Conclusions
The molecular docking assessment of conventional antimalarial drugs as PfGR inhibitors revealed that all drugs exhibited higher affinity than reference inhibitors MD and MB, particularly at the enzyme’s active site. ATV emerged as a promising candidate, displaying the highest affinity for both the active site and the cavity in the interface region, suggesting a dual docking mode. The docking results also showed a moderate correlation with in vitro antiplasmodial activities, suggesting that the potent antimalarial activity of ATV might involve the inhibition of PfGR. ATV and CQ demonstrated a binding mode similar to inhibitor MB at the enzyme’s active site, interacting with the cofactor FADH2 through H-bonds and \(\pi-\pi\) interactions. Valine 383 plays an essential role in assisting the drug binding to the active site, acting as an “anchor” residue and keeping it close to the cofactor FADH2. Electrochemical analysis suggested that ATV and CQ, despite being less efficient than MB, could act as moderate “subversive substrates” at the active site.
Exploring the enzyme’s cavity highlighted distinct binding modes, with ATV and AQ exhibiting notable affinity. Their polar groups of naphtoquinone/quinoline rings play a crucial role as robust H-bond acceptors, while their substituent groups contribute significantly to the ligand’s precise adjustment within site.
Moreover, according to molecular mechanism analysis, dual drugs combining aminoquinoline derivatives and GR inhibitors exhibited significantly greater binding affinity than the reference MD. Results provided insights into their interaction mechanisms, where the presence of the aliphatic ester bond (linker) is essential for effective binding with the enzyme’s active site. Analysis of protein structural dynamics indicated reduced freedom of protein residues when interacting with the dual drugs 1f and 3c, highlighting their effectiveness in stabilizing the protein structure. The linker ester with a long chain does not play an important role in the dual drug binding to the enzyme’s cavity; nonetheless, it provides flexibility for both terminal drugs to interact strongly with the interface region and better fit the enzyme shape. Overall, the study confirms the strong affinity of these dual drugs for PfGR, with stable binding poses maintained throughout the molecular dynamics simulations. In summary, 1f showed to be a good alternative as a dual drug due to the good binding modes reported for both enzyme sites. The study encourages further experiments to investigate the role of PfGR or other disulfide reductases in the mechanisms of action of conventional antimalarial drugs and dual drugs and their contribution to antiparasitic efficacy.
Data Availability
No datasets were generated or analysed during the current study.
References
Rich SM, Ayala FJ (2006) Evolutionary origins of human malaria parasites. Malaria: Genetic and Evolutionary Aspects. Springer, US, Boston, MA, pp 125–146
Singh B, Daneshvar C (2013) Human infections and detection of Plasmodium knowlesi. Clin Microbiol Rev 26:165–184. https://doi.org/10.1128/CMR.00079-12
Ta TH, Hisam S, Lanza M et al (2014) First case of a naturally acquired human infection with Plasmodium cynomolgi. Malar J 13:68. https://doi.org/10.1186/1475-2875-13-68
Imwong M, Madmanee W, Suwannasin K et al (2019) Asymptomatic natural human infections with the simian malaria parasites Plasmodium cynomolgi and Plasmodium knowlesi. J Infect Dis 219:695–702. https://doi.org/10.1093/infdis/jiy519
World Health Organization (2023) World malaria report 2023. Licence: CC BY-NC-SA 3.0 IGO. https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2023. Accessed 16 May 2024
Ministério da Saúde (2024) Boletim epidemiológico da malária vol 55: Dia da Malária nas Américas – um panorama da malária no Brasil em 2022 e no primeiro semestre de 2023. https://www.gov.br/saude/pt-br/centrais-de-conteudo/publicacoes/boletins/epidemiologicos/edicoes/2024/boletim-epidemiologico-volume-55-no-01/view. Accessed 16 May 2024
Ministério da Saúde (2024) Boletim epidemiológico da malária vol 51: Dia Mundial da Malária. https://www.gov.br/saude/pt-br/assuntos/saude-de-a-a-z/m/malaria/situacao-epidemiologica-da-malaria/boletins-epidemiologicos-de-malaria/boletim-epidemiologico-volume-51-no-17-2020-dia-mundial-da-malaria/view. Accessed 19 Jan 2024
Cowman AF, Healer J, Marapana D, Marsh K (2016) Malaria: biology and disease. Cell 167:610–624. https://doi.org/10.1016/j.cell.2016.07.055
Biot C, Castro W, Botté CY, Navarro M (2012) The therapeutic potential of metal-based antimalarial agents: implications for the mechanism of action. Dalton Trans 41:6335. https://doi.org/10.1039/c2dt12247b
de Villiers KA, Egan TJ (2021) Heme detoxification in the malaria parasite: a target for antimalarial drug development. Acc Chem Res 54:2649–2659. https://doi.org/10.1021/acs.accounts.1c00154
Vasquez M, Zuniga M, Rodriguez A (2021) Oxidative stress and pathogenesis in malaria. Front Cell Infect Microbiol 11. https://doi.org/10.3389/fcimb.2021.768182
Dubois VL, Platel DFN, Pauly G, Tribouleyduret J (1995) Plasmodium berghei: implication of intracellular glutathione and its related enzyme in chloroquine resistance in vivo. Exp Parasitol 81:117–124. https://doi.org/10.1006/expr.1995.1099
Jortzik E, Becker K (2012) Thioredoxin and glutathione systems in Plasmodium falciparum. Int J Med Microbiol 302:187–194. https://doi.org/10.1016/j.ijmm.2012.07.007
Sarma GN, Savvides SN, Becker K et al (2003) Glutathione reductase of the malarial parasite Plasmodium falciparum: crystal structure and inhibitor development. J Mol Biol 328:893–907. https://doi.org/10.1016/S0022-2836(03)00347-4
Ginsburg H, Famin O, Zhang J, Krugliak M (1998) Inhibition of glutathione-dependent degradation of heme by chloroquine and amodiaquine as a possible basis for their antimalarial mode of action. Biochem Pharmacol 56:1305–1313. https://doi.org/10.1016/S0006-2952(98)00184-1
Huber PC, Almeida WP, de Fátima  (2008) Glutationa e enzimas relacionadas: papel biológico e importância em processos patológicos. Quim Nova 31:1170–1179. https://doi.org/10.1590/S0100-40422008000500046
Buchholz K, Schirmer RH, Eubel JK et al (2008) Interactions of methylene blue with human disulfide reductases and their orthologues from Plasmodium falciparum. Antimicrob Agents Chemother 52:183–191. https://doi.org/10.1128/AAC.00773-07
Morin C, Besset T, Moutet J-C et al (2008) The aza-analogues of 1,4-naphthoquinones are potent substrates and inhibitors of plasmodial thioredoxin and glutathione reductases and of human erythrocyte glutathione reductase. Org Biomol Chem 6:2731. https://doi.org/10.1039/b802649c
Ehrhardt K, Davioud-Charvet E, Ke H et al (2013) the antimalarial activities of methylene blue and the 1,4-naphthoquinone 3-[4-(trifluoromethyl)benzyl]-menadione are not due to inhibition of the mitochondrial electron transport chain. Antimicrob Agents Chemother 57:2114–2120. https://doi.org/10.1128/AAC.02248-12
Belorgey D, Antoine Lanfranchi D, Davioud-Charvet E (2013) 1,4-Naphthoquinones and other NADPH-dependent glutathione reductase- catalyzed redox cyclers as antimalarial agents. Curr Pharm Des 19:2512–2528. https://doi.org/10.2174/1381612811319140003
Iribarne F, González M, Cerecetto H et al (2007) Interaction energies of nitrofurans with trypanothione reductase and glutathione reductase studied by molecular docking. J Mol Struct (Thoechem) 818:7–22. https://doi.org/10.1016/j.theochem.2007.04.035
Iribarne F, Paulino M, Aguilera S, Tapia O (2009) Assaying phenothiazine derivatives as trypanothione reductase and glutathione reductase inhibitors by theoretical docking and Molecular Dynamics studies. J Mol Graph Model 28:371–381. https://doi.org/10.1016/j.jmgm.2009.09.003
Tyagi C, Bathke J, Goyal S et al (2015) Targeting the intersubunit cavity of Plasmodium falciparum glutathione reductase by a novel natural inhibitor: Computational and experimental evidence. Int J Biochem Cell Biol 61:72–80. https://doi.org/10.1016/j.biocel.2015.01.014
Färber PM, Arscott LD, Williams CH et al (1998) Recombinant Plasmodium falciparum glutathione reductase is inhibited by the antimalarial dye methylene blue. FEBS Lett 422:311–314. https://doi.org/10.1016/S0014-5793(98)00031-3
Shibeshi MA, Kifle ZD, Atnafie SA (2020) Antimalarial drug resistance and novel targets for antimalarial drug discovery. Infect Drug Resist 13:4047–4060. https://doi.org/10.2147/IDR.S279433
Tibon NS, Ng CH, Cheong SL (2020) Current progress in antimalarial pharmacotherapy and multi-target drug discovery. Eur J Med Chem 188:111983. https://doi.org/10.1016/j.ejmech.2019.111983
Sullivan DJ, Gluzman IY, Russell DG, Goldberg DE (1996) On the molecular mechanism of chloroquine’s antimalarial action. Proc Natl Acad Sci 93:11865–11870. https://doi.org/10.1073/pnas.93.21.11865
Buller R, Peterson ML, Almarsson Ö, Leiserowitz L (2002) Quinoline binding site on malaria pigment crystal: a rational pathway for antimalaria drug design. Cryst Growth Des 2:553–562. https://doi.org/10.1021/cg025550i
Camarda G, Jirawatcharadech P, Priestley RS et al (2019) Antimalarial activity of primaquine operates via a two-step biochemical relay. Nat Commun 10:3226. https://doi.org/10.1038/s41467-019-11239-0
Davioud-Charvet E, Delarue S, Biot C et al (2001) A prodrug form of a Plasmodium falciparum glutathione reductase inhibitor conjugated with a 4-anilinoquinoline. J Med Chem 44:4268–4276. https://doi.org/10.1021/jm010268g
Biot C, Bauer H, Schirmer RH, Davioud-Charvet E (2004) 5-Substituted tetrazoles as bioisosteres of carboxylic acids. Bioisosterism and Mechanistic Studies on Glutathione Reductase Inhibitors as Antimalarials. J Med Chem 47:5972–5983. https://doi.org/10.1021/jm0497545
Adigun RA, Malan FP, Balogun MO, October N (2022) Rational optimization of dihydropyrimidinone‐quinoline hybrids as Plasmodium falciparum glutathione reductase inhibitors. ChemMedChem 17. https://doi.org/10.1002/cmdc.202200034
Manhas A, Patel A, Lone MY et al (2018) Identification of Pf ENR inhibitors: a hybrid structure-based approach in conjunction with molecular dynamics simulations. J Cell Biochem 119:8490–8500. https://doi.org/10.1002/jcb.27075
Manhas A, Lone MY, Jha PC (2019) Multicomplex-based pharmacophore modeling in conjunction with multi-target docking and molecular dynamics simulations for the identification of Pf DHFR inhibitors. J Biomol Struct Dyn 37:4181–4199. https://doi.org/10.1080/07391102.2018.1540362
Manhas A, Lone MY, Jha PC (2019) In search of the representative pharmacophore hypotheses of the enzymatic proteome of Plasmodium falciparum: a multicomplex-based approach. Mol Divers 23:453–470. https://doi.org/10.1007/s11030-018-9885-5
Macetti G, Loconte L, Rizzato S et al (2016) Intermolecular recognition of the antimalarial drug chloroquine: a quantum theory of atoms in molecules–density functional theory investigation of the hydrated dihydrogen phosphate salt from the 103 K x-ray structure. Cryst Growth Des 16:6043–6054. https://doi.org/10.1021/acs.cgd.6b01069
Semeniuk A, Niedospial A, Kalinowska-Tluscik J et al (2008) Molecular geometry of antimalarial amodiaquine in different crystalline environments. J Mol Struct 875:32–41. https://doi.org/10.1016/j.molstruc.2007.03.065
Rubin JR, Swaminathan P, Sundaralingam M (1992) Structure of the anti-malarial drug primaquine diphosphate. Acta Crystallogr C 48:379–382. https://doi.org/10.1107/S0108270191008831
Skórska A, Śliwiński J, Oleksyn BJ (2006) Conformation stability and organization of mefloquine molecules in different environments. Bioorg Med Chem Lett 16:850–853. https://doi.org/10.1016/j.bmcl.2005.11.016
Malpezzi L, Fuganti C, Maccaroni E et al (2010) Thermal and structural characterization of two polymorphs of Atovaquone and of its chloro derivative. J Therm Anal Calorim 102:203–210. https://doi.org/10.1007/s10973-010-0685-0
Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR (2012) Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J Cheminformatics 4:17. https://doi.org/10.1186/1758-2946-4-17
Neese F (2022) Software update: The ORCA program system—Version 5.0. WIREs Computational Molecular Science 12. https://doi.org/10.1002/wcms.1606
Barone V, Cossi M (1998) Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J Phys Chem A 102:1995–2001. https://doi.org/10.1021/jp9716997
BIOVIA (2021) Dassault Systèmes, BIOVIA Discovery Studio, 21.1.20298, Dassault Systèmes, San Diego
Case DA, Cheatham TE, Darden T et al (2005) The Amber biomolecular simulation programs. J Comput Chem 26:1668–1688. https://doi.org/10.1002/jcc.20290
Salomon-Ferrer R, Case DA, Walker RC (2013) An overview of the Amber biomolecular simulation package. WIREs Comput Mol Sci 3:198–210. https://doi.org/10.1002/wcms.1121
Morris GM, Huey R, Lindstrom W et al (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791. https://doi.org/10.1002/jcc.21256
Pettersen EF, Goddard TD, Huang CC et al (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. https://doi.org/10.1002/jcc.20084
Vassetti D, Pagliai M, Procacci P (2019) Assessment of GAFF2 and OPLS-AA General force fields in combination with the water models TIP3P, SPCE, and OPC3 for the solvation free energy of druglike organic molecules. J Chem Theory Comput 15:1983–1995. https://doi.org/10.1021/acs.jctc.8b01039
He X, Man VH, Yang W et al (2020) A fast and high-quality charge model for the next generation general AMBER force field. J Chem Phys 153. https://doi.org/10.1063/5.0019056
Jorgensen WL, Chandrasekhar J, Madura JD et al (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935. https://doi.org/10.1063/1.445869
Berendsen HJC, Postma JPM, van Gunsteren WF et al (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81:3684–3690. https://doi.org/10.1063/1.448118
Uberuaga BP, Anghel M, Voter AF (2004) Synchronization of trajectories in canonical molecular-dynamics simulations: observation, explanation, and exploitation. J Chem Phys 120:6363–6374. https://doi.org/10.1063/1.1667473
van Gunsteren WF, Berendsen HJC (1977) Algorithms for macromolecular dynamics and constraint dynamics. Mol Phys 34:1311–1327. https://doi.org/10.1080/00268977700102571
Roe DR, Cheatham TE (2013) PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput 9:3084–3095. https://doi.org/10.1021/ct400341p
Miller BR, McGee TD, Swails JM et al (2012) MMPBSA.py : An Efficient Program for End-State Free Energy Calculations. J Chem Theory Comput 8:3314–3321. https://doi.org/10.1021/ct300418h
Wang E, Sun H, Wang J et al (2019) End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem Rev 119:9478–9508. https://doi.org/10.1021/acs.chemrev.9b00055
Yayon A, Cabantchik ZI, Ginsburg H (1984) Identification of the acidic compartment of Plasmodium falciparum-infected human erythrocytes as the target of the antimalarial drug chloroquine. EMBO J 3:2695–2700. https://doi.org/10.1002/j.1460-2075.1984.tb02195.x
De Souza PC, Quadros HC, Aboagye SY et al (2022) A hybrid of amodiaquine and primaquine linked by gold(I) is a multistage antimalarial agent targeting heme detoxification and thiol redox homeostasis. Pharmaceutics 14:1251. https://doi.org/10.3390/pharmaceutics14061251
Friebolin W, Jannack B, Wenzel N et al (2008) Antimalarial dual drugs based on potent inhibitors of glutathione reductase from Plasmodium falciparum. J Med Chem 51:1260–1277. https://doi.org/10.1021/jm7009292
Villareal P, WJ (2017) Síntese e caracterização de complexos de platina (II) com ligandos fosfínicos e cloroquina: estudo de suas interações com o ADN e avaliação de suas atividades citotóxicas. Dissertation. Universidade Federal de São Carlos. https://repositorio.ufscar.br/handle/ufscar/7962?show=full. Accessed 16 May 202
Daniel L, Karam A, Franco CH, Conde C, de Morais AS, Mosnier J, Fonta I, Villareal WJ, Pradines B, Moreira DRM, Navarro M (2023) Metal(triphenylphosphine)-Atovaquone complexes: synthesis, antimalarial activity, and suppression of heme detoxification. [Manuscript submitted for publication]
Müller T, Johann L, Jannack B et al (2011) Glutathione reductase-catalyzed cascade of redox reactions to bioactivate potent antimalarial 1,4-napthoquinones – a new strategy to combat malarial parasites. J Am Chem Soc 133:11557–11571. https://doi.org/10.1021/ja201729z
Basco L, Gillotin C, Gimenez F et al (1992) In vitro activity of the enantiomers of mefloquine, halofantrine and enpiroline against Plasmodium falciparum. Br J Clin Pharmacol 33:517–520. https://doi.org/10.1111/j.1365-2125.1992.tb04081.x
Lanfranchi DA, Belorgey D, Müller T et al (2012) Exploring the trifluoromenadione core as a template to design antimalarial redox-active agents interacting with glutathione reductase. Org Biomol Chem 10:4795. https://doi.org/10.1039/c2ob25229e
Grellier P, Marozienė A, Nivinskas H et al (2010) Antiplasmodial activity of quinones: roles of aziridinyl substituents and the inhibition of Plasmodium falciparum glutathione reductase. Arch Biochem Biophys 494:32–39. https://doi.org/10.1016/j.abb.2009.11.012
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652. https://doi.org/10.1063/1.464913
Hehre WJ, Ditchfield R, Pople JA (1972) Self—consistent molecular orbital methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J Chem Phys 56:2257–2261. https://doi.org/10.1063/1.1677527
Birth D, Kao W-C, Hunte C (2014) Structural analysis of atovaquone-inhibited cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nat Commun 5:4029. https://doi.org/10.1038/ncomms5029
Acknowledgements
The authors thank SDumont/LNCC and NEQC (UFJF) for the computational resources for calculations and D. Quintanilha for helping generate some images. FHCF would additionally like to thank CeMEAI/EULER and Repesq/UFJF for the computational resources and CAPES, CNPq, and FAPEMIG for the scholarships provided.
Funding
This work was supported by the Minas Gerais State Agency for Research and Development FAPEMIG (BPD-00777–22), the National Council for Scientific and Technological Development CNPq, and the Coordination of Superior Level Staff Improvement CAPES (Finance Code 001).
Author information
Authors and Affiliations
Contributions
This study was conceptualized and written by GYSD. FHCF, BAO, LVD, and LRP conducted the molecular dockings. FHCF also performed molecular dynamics calculations and contributed to writing. MN supervised the work and revised the manuscript. All authors approved the final version.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ferreira, F.H.d.C., Pinto, L.R., Oliveira, B.A. et al. Analysis of the interaction of antimalarial agents with Plasmodium falciparum glutathione reductase through molecular mechanical calculations. J Mol Model 30, 181 (2024). https://doi.org/10.1007/s00894-024-05968-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-024-05968-3