
Abstract
Context
In modern searches for the structure of high-energy-density compounds with high operational, detonation, and physicochemical characteristics, a special place belongs to salts, which have a number of significant advantages over neutral compounds. The development of this area of HEDM is hampered by the lack of effective calculation schemes for estimating the enthalpy of formation DHf0 of salts, as a key parameter in assessing the prospects for their use. Based on the author’s method (MICCM), which is superior in accuracy to currently available calculation methods, the enthalpies of formation of various salts of nitrates and perchlorates for a promising class of high-energy amino-1,2,4-triazoles are calculated and the accuracy of calculations is estimated by other methods. Relationships between the thermochemical characteristics of salts depending on various cations are considered. Among the considered compounds, calculations of the enthalpies of salts of three amino-1,2,4-triazoles showed a significant discrepancy with the experimental data.
Methods
Calculations DHf0of salts were performed using three methods: volume-base thermodynamic (Jenkins/Bartlett method), the method of adding of ions contributions (MAIC, Matyushin’s method), and the method of ions and cocrystals contribution mixing (MICCM, Khakimov’s method). Calculations by the MICCM method were carried out on the basis of quantum chemistry methods (when estimating the enthalpies of formation in the gas phase) and the method of atom-atom potentials (AAP) when calculating the enthalpy of sublimation of salts. We have optimized all the structures in the gas phase using the Becke three hybrid exchange and Lee-Yang-Parr correlation functional with Grimme’s dispersion correction, B3LYP-D2, and aug-cc-pVDZ basis set using the Gaussian16 software. The AAP calculations were performed using the FitMEP software packages (for adjusting the charges of the molecular electrostatic potential) and PMC (for the procedure for constructing crystal packings and searching for optimal ones).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The desire to create an “ideal structure” of high-energy compounds with high performance, detonation, and physicochemical characteristics is always relevant for researchers in this field of science. Therefore, the search for the structure of new promising high-energy compounds does not stop [1,2,3,4]. In this regard, salts should be noted, since they have a number of advantages over neutral compounds. For example, they have a lower vapor pressure, a higher density of molecular crystals, and their structure can be varied by changing different ions inside the salts [5,6,7]. As a rule, in the synthesis of high-energy compounds, salts of nitric and perchloric acids are considered among the first because of the high oxygen content in them.
When evaluating the prospects for using the resulting salts, an important task is to determine their thermochemical properties, especially the enthalpy of formation (ΔHf0). This problem can be solved using computer simulations or experimental methods, for example, using combustion calorimetry. However, the determination of the effectiveness of a substance and, above all, the enthalpy of formation before the stage of its synthesis ensures the expediency of its creation. In this regard, the development of computational methods with a high accuracy of estimating ΔHf0 of compounds is relevant, since it is the enthalpy of formation that significantly affects the assessment of performance properties.
For this reason, the purpose of this work was a comparative analysis of the quality of calculation methods in assessing the enthalpies of formation for various nitrate and perchlorate salts and, on this basis, the estimation of ΔHf0 for the salts of a promising class of high-energy amino-1,2,4-triazoles.
Methods
As is known, the most commonly used calculation scheme is called volume-base thermodynamic (Jenkins/Bartlett method) [8, 9]. The VBT method is based on the dependence of the enthalpy of sublimation on the molecular volume of the salt, calculated as the sum of the molecular volumes of the constituent ions of the salt. In this method, the dependence of the lattice energy (Elattice = − UPOT) on the salt molecular volume (Vmol) can be expressed by the equation:
Subsequently, the lattice energy Elattice is converted into the enthalpy of sublimation ΔHsubl(salt), and the enthalpy of salt formation is estimated by the equation:
The corresponding coefficients and the complete calculation procedure are given in the original work [9].
Another lesser known method is the method of adding of ions contributions (MAIC, Matyushin’s method), developed at the Institute of Chemical Physics of the Russian Academy of Sciences by Dr. Yu. Matyushin [10]. In this method, the ΔHf0 value of the salt is obtained by adding the corresponding values of the anion (ΔHan) and the cation (ΔHcat):
The basis of this additive method is an integrated thermochemical cycle, which leads to the division of the enthalpy of salt formation into anionic and cationic parts. Table 1 lists some of the ionic contributions.
So, according to this method (Eq. (3)) ammonium nitrate has a calculated value of the enthalpy of formation in the solid phase equal to 69.75 − 157.12 = − 87.37 kcal mol−1. The experimental value is − 87.38 kcal mol−1 (Table 2). In general, the error of this method when calculating the enthalpies of salt formation does not exceed 1–2 kcal mol−1; however, the application of the method is limited to the values given in the original article for 20 cations and 20 anions [10].
The recently developed new method for estimating the enthalpies of salt formation—the method of ions and cocrystals contribution mixing (MICCM, Khakimov’s method) [11] is based on fundamentally different provisions than the VBT and MAIC methods, it is an ab initio method and therefore allows you to perform an alternative assessment of the quality of calculations specified methods.
The method is based on relationships similar to Eq. (2), except that it is applied to both the neutral and ionic form, and then the value is averaged. So for the cation [AH]+ and anion [B]− and neutral molecules [A] and [HB] the dependence will look like:
Each part in Eq. (4) is calculated by (2):
The composition of the salt is not important and may be different from the 1:1 ratio.
The gas components of Eqs. (5) and (6) are calculated using quantum chemical calculation methods, based on atomization energies and various composite methods [12,13,14] (we used the Gaussian09 software package [15] for this purpose).
The enthalpies of sublimation of [AH]+[B]− salts and [A][HB] cocrystals were estimated by us by the corresponding predictive modeling of crystal lattices according on the atom–atom potentials method and the methodology presented in [16,17,18,19,20], but, of course, other methods can be used for these purposes. The basis of the method is the division of the atom–atom potential interactions into van der Waals and electrostatic components, followed by summation and minimization. For the fitting of charges for electrostatic interaction, program FitMEP [16] was used, and for the procedure for constructing crystalline packings, program PMC [17]. The form of interaction is chosen in the form of potentials “6–12” [21]:
For Z′ = 2, eleven space groups (P21/c, P212121, P-1, P21, Pbca, C2/c, Pna21, Pca21, Cc, C2, P1) cover almost 95% of the known structures of this type (organic cocrystals and salts) [22]. These groups were used in the calculations. Technical details of the calculations are given in the Supplementary Information (SI) section.
To evaluate the accuracy of the methods mentioned above in estimating the enthalpy of formation for perchlorates and nitrates, we used the NIST database [23] and other experimental studies [24,25,26,27] on the determination of the enthalpies of salt formation and collected in Table 2 different cation values.
The average value of the difference between the enthalpies of formation for perchlorates and nitrates ΔHan(ClO4) − ΔHan(NO3) from Table 2 is 19.43 kcal mol−1, which is quite close to the value according to the MAIC method (Table 1):
\({{\Delta H}}_{\mathrm{an}}\left({\mathrm{ClO}}_{4}\right)-{{\Delta H}}_{\mathrm{an}}\left({\mathrm{NO}}_{3}\right)= - {140.43\hspace{0.17em}+\hspace{0.17em}157.12\hspace{0.17em}=\hspace{0.17em}16.69\mathrm{ kcal\; mol}}^{-1}\)
Depending on the data source, the tabular values may differ slightly from each other, which leads to an almost imperceptible change in the average value, which determines the applicability of estimating the enthalpies of formation by additive methods.
Earlier this position was noted for nitrates and salts of dinitramic acid [24, 28] in the form ΔHan(N(NO2)2) − ΔHan(NO3) = 55 kcal mol−1, which also follows from the MAIC method:
\({{\Delta H}}_{\mathrm{an}}\left({\mathrm{N}\left({\mathrm{NO}}_{2}\right)}_{2}\right)-{{\Delta H}}_{\mathrm{an}}\left({\mathrm{NO}}_{3}\right)=-{\hspace{0.17em}=\hspace{0.17em}\hspace{0.17em}-\hspace{0.17em}101.91\hspace{0.17em}+\hspace{0.17em}157.12\hspace{0.17em}=\hspace{0.17em}55.21\mathrm{ kcal\; mol}}^{-1}\)
Results and discussion
Based on the methodology and results for nitrate salts obtained by us in [11], the enthalpies of formation for a number of organic and inorganic salts of perchlorates were estimated (Table 3). As can be seen from this table, the enthalpies of formation agree fairly well with the experimental values.
Due to the existing limitations on the atomic composition (the set of atom–atom potentials is limited by C, H, N, O atoms), when determining the enthalpies of sublimation, the values for perchlorates in calculations by the MICCM method were taken from the estimate of nitrates by the MAIC method according to Eqs. (3) and (4).
It is clearly seen from Table 3 that the MAIC method gives excellent agreement with experiment within 1–2 kcal mol−1. The MICCM values are somewhat worse and the differences from the experiment are up to 3–4 kcal mol−1. However, the advantage is that you can get values for new salts and add values for ions using the MAIC method, since the number of available values for MAIC is very limited. The mean absolute errors for MICCM and MAIC from Table 3 are 2.2 kcal mol−1 and 0.96 kcal mol−1, respectively.
The VBT method gives a significant difference from the experimental values, which for nitrates is about 15–20 kcal mol−1 or more, depending on the method for calculating the molecular volume (in Table 3, method CBS-4M, Monte-Carlo DFT/6-31G(d,p)).
Note that the value for the enthalpy of formation (MICCM) of diaminoguanidinium perchlorate was predicted for the first time.
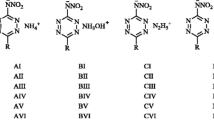
Convinced of the reliability of calculations by the MICCM and MAIC methods, we estimated the values of the enthalpies of formation of salts of amino-1,2,4-triazoles, for which there were experimental data on the enthalpies of formation of their nitrates and perchlorates [29] (Fig. 1). In [30], the same authors presented other values, which are also included in Table 4.

The structure of the considered salts of amino-1,2,4-triazoles
Three methods were used to estimate the enthalpy of formation in gas in Eq. (2) for VBT and Eqs. (5) and (6) for MICCM: CBS-4M, CBS-QB3, G3B3 [13, 14].
It is known [31] that Gn methods are more accurate than CBS, but require much more computational resources. Methane, ethane, benzene, ammonia, hydrazine, and water were chosen to evaluate the quality of quantum chemical methods for the test set when calculating the enthalpy of formation in gas. The average deviation of the calculated absolute values from the experimental ones for CBS-4M, CBS-QB3, and G3B3 was 2.08 kcal mol−1, 0.76 kcal mol−1, and 0.52 kcal mol−1, respectively. The largest error in CBS-4M calculations corresponds to aromatic compounds. As stated above from [31] and our test set calculations, we chose the G3B3 method as the most accurate.
The enthalpies of formation were calculated according to two schemes: by direct calculations using the energy atomization method and using isodesmic reactions (Table 4). The latter method was used to rule out systematic errors. In total, six isodesmic reactions were considered for cations (R′ = H, CH3) and neutral aminotriazoles (R′ = H, CH3). Isodesmic reactions for cations are presented in Scheme 1. The scheme for neutral compounds is not shown due to the identity of the reactions.

Isodesmic reactions for cations of amino-1,2,4-triazoles
Unlike VBT, for MICCM it is necessary to calculate the neutral complex in the gas phase and therefore, similarly to Scheme 1, the enthalpies of formation in gas for neutral aminotriazoles, as well as for the corresponding isodesmic reactions, were calculated using the experimental data of the compounds involved in isodesmic reactions: ammonia, hydrazine, methane, methylamine, and 1H-1,2,4-triazole (Table 4).
An algorithm for calculating the enthalpies of formation based on isodesmic reactions and “direct” quantum chemical methods, as well as information on modeling the crystal structure of the compounds under consideration, are presented in the SI. In Tables 4 and 5, DM corresponds to calculations by the direct method, and IM1 by the method of isodesmic reactions for R′ = H and IM2 for isodesmic reactions with R′ = CH3.
One can see almost complete identity of the values of the enthalpies of formation in the gas phase for cations and for neutral aminotriazoles calculated both by direct and isodesmic methods. The obtained values of CBS-4M and G3B3 are also quite close to each other.
An alternative way to check the obtained value of the formation enthalpy of 3-amino-1,2,4-triazole in the gas phase is to solve the inverse problem according to Eq. (2). The enthalpy of formation of 3-amino-1,2,4-triazole in the solid phase is known [32] and is equal to 18.36 kcal mol−1. We simulated its crystal structure and determined the enthalpy of sublimation, which turned out to be 27.7 kcal mol−1. Thus, it is definitely possible to estimate the enthalpy of this compound in the gas phase as 46.06 kcal mol−1, which corresponds to the correctness of quantum chemical calculations (40.68–45.29 kcal mol−1).
The performed calculations indicate that a possible error in determining the “gas part” of Eqs. (2), (5), and (6) is excluded.
Tables 6 and 7 show the formation enthalpies of nitrates and perchlorates for 1-amino, 3-amino, and 4-amino-1,2,4-triazoles, obtained by the VBT and MICCM methods. The direct method for estimating the enthalpies in the gas phase by various methods of quantum chemistry was used in the calculations.
The strong difference between the given values of the enthalpies of formation for compounds 1, 3, 4, and 6 is striking.
It should be noted that the VBT method overestimates the MICCM + MAIC method by an average of 23 kcal mol−1 for nitrates and 13 kcal mol−1 for perchlorates, depending on the calculation method used for the gas phase. There are known deviations of the results obtained by the VBT method from the experimental values of more than 150 kcal mol−1 [33].
The difference between the experimental values for nitrates 1 and 3 from the MICCM (G3B3) values is 13.5 kcal mol−1 and − 23.5 kcal mol−1 for the data from [29], and 22.6 kcal mol−1 and 21.2 kcal mol−1 for the data from [30], respectively. For 2, the difference is only 2.8 kcal mol−1. For salts 1 and 3, the difference is significant and often with the opposite sign.
The difference between the given experimental values [29] for nitrates and perchlorates of 1-amino and 4-amino-1,2,4-triazoles is:
ΔHf0(4) − ΔHf0 (1) = ΔHan(ClO4) − ΔHan(NO3) = 77.05 kcal mol.−1
ΔHf0(6) − ΔHf0 (3) = ΔHan(ClO4) − ΔHan(NO3) = 97.57 kcal mol.−1
Such a difference in the enthalpies of formation for perchlorates and nitrates strongly disagrees with the MAIC value of 16.69 kcal mol−1 and, in our opinion, the values of the enthalpies of formation of salts of amino-1,2,4-triazoles should be revised.
Crystal packings with minimum lattice energies for salts 1 and 2 with space groups P-1 and Pca21 are shown in Fig. 2. We did not find experimental X-ray diffraction data for these compounds.

Model crystal packings of 1 (left) and 2 (right)
However, the Cambridge Structural Database has an X-ray diffraction pattern for salt 3. The model packing with minimum energy (P21/c) differs from the experimental one (group Cc). The minimum in the gas phase for the 4-amino-1,2,4-aminotriazolium cation corresponds to symmetry Cs where the amino group is perpendicular to the plane of the triazole ring. In the experimental structure, the amino group is somewhat rotated (~ 26°) relative to the plane of the ring with point group C1 (Fig. 3).

Experimental (left) and simulated model (right) of crystal packings of 3
The difference between the lattice energy for the global minimum structure and the packing observed in the experiment is about 5 kcal mol−1. Taking into account the averaging of the ionic and neutral parts in Eq. (4), the difference is reduced to 2.5 kcal mol−1, which is quite acceptable for estimating the enthalpy of formation. A list of the 10 lowest energy polymorphs for each of the salt and the corresponding cocrystal is given in SI.
The paper [34] presents experimental data on the 1-methyl derivative of compounds 4 and 6, for which the enthalpies of formation were obtained − 5.5 ± 2.4 kcal mol−1 for nitrate and 17.21 ± 4.3 kcal mol−1 for perchlorate, respectively. Without taking into account inaccuracies, the difference between perchlorate and nitrate is 11.71 kcal mol−1 and, taking into account the maximum spread of 18.41 kcal mol−1 of the MAIC method, is well within its accuracy.
We suggest using the following values to calculate cationic contributions for amino-1,2,4-triazoles: for 1-amino-1,2,4-triazolium 151.87 kcal mol−1, for 3-amino-1,2,4-triazolium 113.42 kcal mol−1 (116.22 kcal mol−1 from [10]), and for 4-amino-1,2,4-triazolium 154.39 kcal mol−1. The enthalpies of formation for nitrate and perchlorate salts correspond to the MICCM (G3B3) values from Tables 6 and 7.
The article [35] presents experimental results on the enthalpies of combustion and formation of azobistetrazole-1,1′-oxide salts. Among these salts are salts with guanidine and 4-amino-1,2,4-triazolium cations. On the basis of these two salts, we can carry out an additional verification of the correctness of our results, namely, to calculate the difference in their enthalpies of formation and calculate the contribution from 4-amino-1,2,4-triazolium. The enthalpy of formation of diguanidinium azobistetrazole-1,1′-oxides is 171.42 kcal mol−1, and the enthalpy of formation of di-4-amino-1,2,4-triazolium azobistetrazole-1,1′-oxides is 348.37 kcal mol−1. This means that the difference between the contributions from these two cations is (348.37 − 171.42)/2 = 88.48 kcal mol−1. Taking into account the value of the contribution of guanidinium 65.18 kcal mol−1 (Table 1), we find that for 4-amino-1,2,4-triazolium this value will be about 65.18 + 88.48 = 153.66 kcal mol−1. The value of this estimate coincides very closely with the value of our contribution of 154.39 kcal mol−1.
Below (Table 8) is a summary table for various salts of amino-1,2,4-triazoles using the obtained cationic contributions.
Conclusion
Thus, based on a combination of quantum chemistry methods and atom–atom potentials, the crystal structure for sets of salts of nitric and perchloric acids was simulated and their enthalpy of sublimation was estimated for subsequent use in calculations the enthalpies of salt formation in the solid phase using the recently developed MICCM method. Relationships between the enthalpies of formation of salts of nitric and perchloric acids with the same cations are revealed. Using ammonium nitrate, hydrazine, and various aminoguanidines, the effectiveness of the three most commonly used methods for calculating the enthalpies of salt formation is evaluated. The enthalpy of formation of diaminoguanidine perchlorate (− 20.89 kcal mol−1) was obtained for the first time. For nitrates of 1-amino- and 4-amino-1,2,4-triazoles, significant discrepancies between the calculated and experimental data were revealed. For the formation enthalpy of 3-amino-1,2,4-triazole nitrate, the results of the calculation by the MICCM + MAIC method correspond to the experimental results. Attention is drawn to the significantly overestimated experimental value of the enthalpies of formation for perchlorate salts of amino-1,2,4-triazoles in comparison with the results of calculations performed by all calculated methods, which calls into question the experimental data.
References
Yao W, Xue Y, Quia L, Yang H, Cheng G (2021) Combination of 1,2,3-triazole and 1,2,4-triazole frameworks for new high-energy and low-sensitivity compounds. Energetic Materials Frontiers 2:131–138
Rudakov GF, Sinditskii VP, Andreeva IA, Botnikova AI, Veselkina PR, Konstakyan SK, Yudin NV, Serushkin VV, Cherkaev GV, Dorofeeva OV (2022) Energetic compounds based on a new fused bis[1,2,4]triazolo[1,5-b;5’,1’-f]-1,2,4,5-tetrazine. Chem Eng J 450:138073
Mathpati RS, Yadav AK, Ghule VD, Dharavath S (2022) Potential energetic salts of 5,5’-methylenedi(4H–1,2,4-triazole-3,4-diamine) cation: synthesis, characterization and detonation performance. Energetic Materials Frontiers 3:90–96
Chaplygin DA, Larin AA, Muravyev NV, Meerov DB, Kosareva EK, Kisilev VG, Pivkina AN, Ananyev IV, Fershtat LL (2021) Nitrogen-rich metal-free salts: a new look at the 5-(trinitromethyl)tetrazolate anion as an energetic moiety. Dalton Trans 50:13778–13785
Bayat Y, Taheripouya G (2018) Synthesis of novel energetic N-(1-carboxymethyl-1H-tetrazole-5-yl)-hydrazinium salts. Cent Eur J Energ Mater 15:420–434
Bauer L, Benz M, Klapötke TM, Pignot C, Stierstorfer J (2022) Combining the most suitable energetic tetrazole and triazole moieties: synthesis and characterization of 5-(1-hydroxy-3-nitro-1,2,4-triazol-5-yl)-1-hydroxy-tetrazole and its nitrogen-rich ionic derivatives. Mater Adv 3:3945–3951
Gettings ML, Byrd EFC, Zeller M, Piercey D (2021) Methyl sydnone imine and its energetic salts. New J Chem 45:2228–2236
Mallouk TE, Rosenthal GL, Muller G, Busasco R, Bartlett N (1984) Fluoride ion affinities of germanium tetrafluoride and boron trifluoride from thermodynamic and structural data for (SF3)2GeF6, ClO2GeF5, and ClO2BF4. Inorg Chem 23:3167–3173
Jenkins HDB, Tudela D, Glasser L (2002) Lattice potential energy estimation for complex ionic salts from density measurements. Inorg Chem 41:2364–2367
Matyushin YuN, Kon´kova TS, (2014) The method for assessment of thermochemical properties for salt compounds. Gorenie i Vzryv [Combustion and Explosion] 7:277–287
Khakimov DV, Pivina TS (2022) A new method for predicting the enthalpy of salt formation. J Phys Chem A 126:5207–5214
Klapotke TM (2012) Computational methods, in: Chemistry of high energy materials, Walter de Gruyter, Berlin–Boston, 89.
Montgomery JJA, Frisch MJ, Ochterski JW, Petersson GA (2000) A complete basis set model chemistry. VII. Use of the minimum population localization method. J Chem Phys 112:6532–6542
Curtiss LA, Raghavachari K, Redfern PC, Rassolov V, Pople JA (1998) Gaussian-3 (G3) theory for molecules containing first and second-row atoms. J Phys Chem 109:7764–7776
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich A, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ (2016) Gaussian 09, revision D.01, Gaussian, Inc., Wallingford CT.
Dzyabchenko AV (2008) A multipole approximation of the electrostatic potential of molecules. Russ J Phys Chem A 82:758–766
Dzyabchenko AV (2008) From molecule to solid: the prediction of organic crystal structures. Russ J Phys Chem A 82:1663–1671
Khakimov DV, Dzyabchenko AV, Pivina TS (2019) Crystal structure prediction of bifurazano[3,4-b:3’,4’-f]furoxano[3",4"-d]-oxacycloheptatriene (BFFO) in the experimentally known monohydrated and proposed anhydrous forms. Propellants, Explos, Pyrotech 44:1528–1534
Khakimov DV, Zelenov VP, Baraboshkin NM, Pivina TS (2019) The unusual combination of beauty and power of furoxano-1,2,3,4-tetrazine 1,3-dioxides: a theoretical study of crystal structures. J Mol Model 25:107
Khakimov DV, Fershtat LL, Pivina TS, Makhova NN (2021) Nitrodiaziridines: unattainable yet, but desired energetic materials. J Phys Chem A 125:3920–3927
Momany FA, Carruthers LM, McGuire RF, Scheraga HA (1974) Intermolecular potentials from crystal data. III. Determination of empirical potentials and application to the packing configurations and lattice energies in crystals of hydrocarbons, carboxylic acids, amines, and amides. J Phys Chem 78:1595–1620
Cruz-Cabeza AJ, Pidcock E, Day GM, Motherwell WDS, Jones W (2007) Space group selection for crystal structure prediction of solvates. CrystEngComm 9:556–560
Chase MW (1998) NIST-JANAF thermochemical tables, American Institute of Physics, 4th Edition 1–1952.
Kon´kova TS, Matyushin YuN, Miroshnichenko EA, Vorob´ev AB, (2009) Thermochemical properties of dinitramidic acid salts. Russ Chem Bull 58:2020–2027
Gilliland AA (1962) Heat of formation of nitronium perchlorate. Journal of Research of the National Bureau of Standards-A. Physics and Chemistry 66:447–449
Markowitz MM, Harris RF, Stewart H Jr (1959) The heat of formation of anhydrous lithium perchlorate. J Phys Chem 63:1325–1326
Rafeev VA, Rubtsov YI (1993) Kinetics and mechanism of thermal decomposition of hydroxylammonium nitrate. Russ Chem Bull 42:1811–1815
Wingeborg N, Latypov NV (2003) Triaminoguanidine dinitramide, TAGDN: synthesis and characterization. Propellants, Explos. Pyrotech 28:314–318
Xue H, Arritt SW, Twamley B, Shreeve JM (2004) Energetic salts from N-aminoazoles. Inorg Chem 43:7972–7977
Shreeve JM (2007) Ionic liquids as energetic materials. Report AFRL-SR-AR-TR-07–0094.
Pokon EK, Liptak MD, Feldgus S, Shields GC (2001) Comparison of CBS-QB3, CBS-APNO, and G3 predictions of gas phase deprotonation data. J Phys Chem A 105:10483–11087
Williams MM, McEwan WS, Henry RA (1957) The heat of combustion of substituted triazoles, tetrazoles and related high nitrogen compounds. J Phys Chem 61:261–267
Kon´kova TS, Matyushin YuN, Miroshnichenko EA, Makhov MN, Vorob´ev AB, Inozemtsev AV, (2018) Energetic properties of derivatives of 1,2,4-triazole. Gorenie i Vzryv [Combustion and Explosion] 11:90–99
Schaller U, Keicher T, Weiser V, Krause H, Schlechtriem S (2010) Characterization and combustion of triazolium based salts. In Insensitive munitions and energetic materials technology symposium, Munich, Germany, October 11–14.
Zhang Z, Zhang J, Gozin M (2018) Nitrogen-rich salts based on 1,1’-dihydroxy-5,5’-azobistetrazole: a new family of energetic materials with promising properties. ChemistrySelect 3:3463–3473
Author information
Authors and Affiliations
Contributions
DK did the calculations and came up with the concept of the work and wrote the original text. TP reviewed the work, wrote the conclusions, and participated in the discussion of the work.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Khakimov, D.V., Pivina, T.S. Is everything correct? The formation enthalpy estimation and data revision of nitrate and perchlorate salts. J Mol Model 29, 75 (2023). https://doi.org/10.1007/s00894-023-05477-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-023-05477-9