
Abstract
In the present research, the cation–π interactions in acetaminophen-M complexes (M = Mn+, Fe2+, Co+, Ni2+, and Cu+) are investigated using density functional theory (DFT/ωB97XD) in the gas phase and solution. The results show that the absolute values of energy are reduced in going from the gas phase to the solution. Based on the obtained data, the complexes in water are the most stable. The natural bond orbital (NBO) and the atoms in molecules (AIM) analyses are also applied to achieve more details about the nature of interactions. These results are useful for understanding the role of the drug-receptor interactions in the complexes. According to AIM outcomes, the cation–π interactions are the closed-shell and may indicate the partially covalent nature in the complexes. A comprehensive analysis is also performed on the conceptual DFT parameters of the complexes to evaluate their electronic properties. Our findings show increasing the stability and decreasing the reactivity of the complexes in the solution phase with respect to the gas phase. These interactions are ubiquitous in biological systems, and their importance in theoretical models led us to study such important interactions. The results of this study may be useful for the design and synthesis of a variety of supramolecular complexes with the desired properties.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acetaminophen (also known as Tylenol) is the most commonly taken analgesic worldwide [1,2,3]. It is a p-aminophenol derivative that is used for two main purposes. The first is as a non-opioid analgesic for mild to moderate pain. The second is as an antipyretic to lower fever [4,5,6]. There is some evidence that acetaminophen (AC) may have beneficial effects on blood glucose level and muscle function [7]. As it has antioxidant benefits, it may be useful for protecting the health of the heart and brain [8]. AC gastrointestinal side effects are much less likely than the other drugs, but overdose can lead to fatal liver damage [9]. However, it is not an anti-inflammatory drug, so it does not help reduce swelling or inflammation [10]. The COX, or cyclooxygenase, is an enzyme responsible for the synthesis of prostaglandins [11]. Prostaglandins are chemicals that cause inflammation and swelling. One theory is that the AC increases the pain threshold by inhibiting two isoforms of cyclooxygenase, COX-1 and COX-2. It prevents the synthesis of prostaglandins in the central nervous system, but it has no an anti-inflammatory effect on peripheral nerves [12, 13].
Cation–π interactions indicate the non-covalent attraction between cations and π-systems [14,15,16,17]. In fact, such an interaction occurs between a cation and the permanent quadrupole moment of an aromatic ring. These interactions play a major role in several fields of modern chemistry, especially in the field of supramolecular chemistry and molecular recognition [18,19,20,21,22,23]. The electrostatic and dispersion forces and charge-transfer/inductive effects are factors that are assumed to contribute in the cation–π interactions. The theoretical studies also show several features for the cation–π interactions [24]: (1) the predominant contribution of the polarization term in the cation–π complexes, (2) the reduction of the aromaticity of the ring by cation–π interactions, and (3) the non-participation of the cation atomic orbitals in the π-type molecular orbitals of the complexes.
The nature and energy of cation–π interactions have been investigated in many studies [25,26,27,28,29,30,31]. In 2019, Mohammadi et al. [32] performed DFT calculations on all the complexes formed by interaction between mono- and divalent metal cations (Li+, Na+, K+, Be2+, Mg2+, and Ca2+) with acetaminophen. Investigating the structural and electronic effects of alkali metal and alkaline earth cations with phenylalaninal π-system in the gas phase and solution was done by Alirezapour et al. [33]. In 2020, the cation–π interactions between the anticancer drug altretamine and some mono- and divalent metal cations, such as Li+, Na+, K+, Be2+, Mg2+, and Ca2+, were investigated to evaluate the stability and electronic properties of the complexes [34]. Mohammadi et al. [35] conducted a comprehensive study on the non-covalent interactions of the acetaminophen complex in the presence of various solvents. Also, by Alirezapour et al. [36], the quantum chemical calculations were performed to investigate the effect of cation–π interactions on the structural and electronic characterization of the para-aminosalicylic acid complexes in 2020.
The pharmacology of metal-drug interactions may depend on the method of management; a metal-drug complex can be directed as a therapeutic agent, or the drug can be administered and interact with metal ions in the body. Due to the application of acetaminophen as an important biological molecule chelating with metal ions or change in the polarity of the medium may affect its chemical and biological properties [37, 38]; hence, investigation of its interactions in both the gas phase and the solution is necessary. In this work, the cation–π interactions are modeled by the Mn+, Fe2+, Co+, Ni2+, and Cu+ cations and AC as the π-system. The selection of these ions is mainly due to the important role that they play in many biochemical processes [39,40,41]. Since the explored cations are chosen to have a closed-shell electronic configuration, so they are preferred to be monovalent or divalent, because higher oxidation states can lead to very different results. In this research, the spin multiplicity of the complexes is considered to be the singlet; thus. they are the low spin.
Our goal is to investigate the role of cation–π interactions in the energetic, geometric, spectroscopic, topological, and molecular orbital descriptors of AC complexes. The solute–solvent interactions are responsible for the significant changes in the chemical and physical characteristics of the solute in going from gas phase to solvent phase [42, 43]. In this regard, we have studied the influence of solvents on all complexes. For this purpose, the DFT calculations are used and the AIM and NBO analyses are performed. The harmonic oscillator model of aromaticity (HOMA) index is also applied to provide better insights into the electron delocalization and molecular aromaticity properties in the studied complexes. Finally, a comprehensive analysis is performed on the conceptual DFT parameters of the complexes to evaluate their electronic properties.
Quantum chemical calculations
The quantum chemistry package GAMESS [44] is utilized for all computations in the present study. The geometry of the complexes is optimized within the framework of density functional theory (DFT/ωB97XD) [45] with the 6-311++G(d,p) basis set [46]. This method includes the long-range correction with empirical atom–atom dispersion [47,48,49]; therefore, it is a suitable implement for the analysis of non-covalent complexes. The vibrational frequencies are computed on the optimized geometries at the same level of theory. To describe the interaction between the metal cations with the AC molecule, the binding energy (∆Eion-π) is calculated by the following equation:
where Ecation-π is the total energy of complex, Eπ-system is the energy of AC monomer, and Ecation is the energy of metal ions. The counterpoise method of Boys and Bernardi is employed to compute the basis set superposition error (BSSE) [50]. The self-consistent reaction field (SCRF) method with the polarized continuum model (PCM) is used to investigate the effect of the solvent [51, 52]. The stabilization energy (Estab) is estimated from the total energies to explore the stability of complexes according to the following equation:
where Etot (solvent) and Etot (gas) are the total energies of the complexes in the solvent and gas phase, respectively. The Bader’s quantum theory of atom in molecule (QTAIM) [53] is applied to understand the properties of chemical bonds in the considered complexes. The AIM analysis is implemented using the AIM2000 package [54]. The population analysis by the natural bond orbital (NBO) method [55] is performed on structures optimized at the ωB97XD/6-311++G(d,p) level of theory. The electronic properties of complexes are estimated by the frontier molecular orbitals (FMOs), i.e., the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). Finally, the aromaticity of the AC ring is assessed by the harmonic oscillator model of aromaticity (HOMA), as a geometry-based index [56]. In this investigation, the Ropt and αare obtained at the ωB97XD/6-311++G(d,p) level of theory (for CC bond: Ropt,CC = 1.393 Å and αCC = 89.34).
Results and discussion
Energies
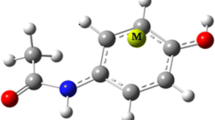
Figure 1 illustrates the structure of AC∙∙∙M ionic complexes with M = Mn+, Fe2+, Co+, Ni2+, and Cu+. The binding energies (∆Eion-π) obtained in the gas phase and the selected solvents are reported in Table 1. As can be seen in this table, the binding energy of the complexes changes appreciably when the solvent effect is applied. According to the ∆Eion-π values calculated for all complexes, it is apparent that the cation–π interaction strength in the gas phase is larger than that in solution. Among the solution phase, the non-polar solvent (chloroform) has a stronger interaction compared to the polar solvent (water). For example, the binding energy of the Fe2+ complex at the ωB97XD/6-311++G(d,p) level using the PCM method drops from − 214.73 kcal mol−1 in the gas phase to − 51.88 kcal mol−1 in the chloroform and to − 19.71 kcal mol−1 in the water. Our findings display that the Ni2+ complex in the gas phase has the highest absolute value of ∆Eion-π. Unlike the gas phase, the maximum value of ∆Eion-π in the solution phase is predicted to be Mn+ complex (see Table 1).

The model representation for the AC∙∙∙M complexes, with M = Mn+, Fe2+, Co+, Ni2+, and Cu+ from (a) front view and (b) top view
The values of total energy (Etot), stabilization energy (Estab), and relative energy (Erel) for all the designated complexes are given in Table 1. Solvents play an important role in the stability of complexes because they are able to change the polarity of the environment. The obtained results reveal that the complexes in water solution are the most stable. The stability order of the complexes is as follows: Fe2+ > Ni2+ > Mn+ > Co+ > Cu+. A similar trend is also observed in the chloroform. As can be seen in Table 1, this stabilization enhances with increasing polarity of the solvent. The relative energies (Erel) are also calculated from the difference between the Etot of complexes in the gas phase and the CHCl3 relative to the water (as the most stable complexes). Figure 2 illustrates chart of the stability order of the complexes in terms of the relative energy in different media. As shown in this figure, the AC complexes with the lowest absolute amount of binding energy in the water solvent have the highest stability in this solvent compared to the others. This means that the solvent plays a key role in the stability of these complexes.

Chart of the stability order in terms of relative energy (Erel) in different media
In order to gain more insight into the properties of the selected complexes, the thermodynamic parameters of Gibbs free energy (ΔG), enthalpy (ΔH), and entropy (ΔS) are calculated at room temperature (298.15 K) and one atmospheric pressure. The results are listed in Table 1. As can be seen in this table, the values of ΔG and ΔH of the complexes are positive and negative, respectively. Since the changes in Gibbs free energy are positive, this implies that the formation of the complexes is not a spontaneous process. The calculated values of ΔH in Table 1 also show that the formation process of all complexes is enthalpically favored (exothermic). Hence, ΔS < 0 and ǀΔHǀ < ǀTΔSǀ. This means that the entropic factor controls the stability of the complexes in both the gas phase and in solution. Therefore, all the thermodynamic properties are not only dependent on the nature of the different cations but are also affected by environmental factors.
Geometric parameters
Table 2 shows the structural parameters of the considered complexes in the gas phase and solutions. The values of dion-π (the distance between cations and the center of the aromatic ring) are important descriptors of the cation–π interactions strength. It can be stated that complexes with the most ǀ∆Eion-πǀ value have the least interaction distance (dion-π) and vice versa. Based on the obtained data, these results can be observed in the monovalent complexes. For example, the Cu+ complex with the largest value of dion-π has the smallest value of ∆Eion-π, while the Mn+ complex with the highest value of ∆Eion-π has the lowest value of dion-π. However, a similar correlation cannot be detected between these values for the Ni2+ and Fe2+ divalent complexes.
The C = C bond length (dC=C) of the AC complexes also changes in the presence of cation-π–interactions. The results of Table 2 show that in most cases, for each ion group (mono- or divalent), the dC=C increase is accompanied with enhancing the ǀ∆Eion-πǀ values. The data obtained in the gas and chloroform phases display that the maximum value of dC=C is related to the Ni2+ complex, while the minimum value corresponds to the Cu+ complex. This indicates that the cation–π interaction in these phases is the strongest/weakest for the Ni2+/Cu+ complexes, respectively. According to the found results, the dC=C values of the complexes decrease on going from the gas phase to the solution, so that the lowest value is observed in the water solvent. This confirms the weaker cation-π interaction of complexes in the solution phase with respect to the gas phase.
The geometric changes created by the cation–π interactions are in line with the spectroscopic data. The vibrational frequencies of the ion-π (ʋion-π) for AC complexes are shown in Table 2. It is prominent that the stronger cation–π interaction causes the vibrational frequencies (ʋion-π) to shift to the higher values. The data in Table 2 indicate that the relationship between ∆Eion-π and ʋion-π is consistent with the studied complexes in the gas phase; but for the complexes of solution phase, these results are only valid for the monovalent ones. The obtained results also show that in most cases, the largest and smallest shifts belong to the gas phase and solution complexes, respectively.
Aromaticity analysis
The geometric parameters of the complexes may also be applied to describe π-electron delocalization [57]. One of these descriptors is the geometry-based aromaticity index [58]. In this work, the HOMA index [56] is selected to explain the molecular aromaticity properties in complexes and is defined according to the following:
where, n is the number of bonds studied, α is a normalization constant, Ropt is the optimal value of bond lengths for ideally aromatic systems and Ri stands for bond lengths taken into consideration. The HOMA is zero for a model non-aromatic system and it is equal to 1 for full aromatic systems.
The results of Table 2 show that in most cases, the calculated values of HOMA are close to the ideal aromaticity index (HOMA = 1). Our findings in all phases also exhibit that the Cu+ complex has the greatest aromaticity, while the least aromaticity is related to the Ni2+ complex. In other words, the Ni2+/Cu+ complexes have less/more effect on the improvement of the aromaticity, respectively. These results prove that the aromaticity depends on the type of the cation. It can be seen in Table 2 that the values of HOMA enhance as the dielectric constant increases (except for Ni2+ complex).
After optimization of the Ni2+ complex (in the gas phase), it is observed that the Ni2+ cation is connected to the oxygen atom of the carbonyl group and its structure deviates from the planar state. This may be due to the effects of electrostatic attraction between the Ni2+ ion and the electronegative oxygen atom. In addition, in the Ni2+ complex, due to the strong cation–π interaction, a distorted architecture of the “binary-system moiety” is created in this structure, which causes a little bit the aromaticity of the benzene ring to be lost. This makes the cation–π interaction less efficient in this complex [59, 60] and reduces the aromaticity of the benzene ring relative to the others.
The dipole moment (μ°) values in the gas phase and the titled solvents are presented in Table 2. As can be seen, the dipole moment of the complexes increases on going from the gas phase to the solution, so that the greatest value is observed in the water solvent. Since, the dipole moment of different complexes depends on the polarity of the medium, hence, their stability may be affected by such changes. Our results show that the complexes become more stable due to their large dipole moments. The calculated dipole moments in the solution phase are in the range of 8.06 D–12.77 D and the maximum and minimum values of μ° correspond to the Fe2+ and Ni2+ complexes, respectively. Intermolecular interactions in materials are also regulated by dipole moments. For polar solvents, intermolecular interactions decrease as dipole moments increase.
Polarizability is the second derivative of the energy relating to an electric field [61]. The cation–π interactions strongly depend on the molecular polarizability values of the aromatic compounds [62]. The data obtained in Table 2 show that the maximum and minimum values of polarizability belong to the solution and gas phases, respectively. This means that the complexes produce the weaker interactions (ΔEion-π) in the solution phase with respect to the gas phase. In addition, the high values of the polarizability suggest that the contributions of electrostatic and the dispersion play an important role in the formation energy of the interacting species [63]. A direct relationship between the dipole moment and the polarizability of the complexes can be observed both in the gas phase and in solution. Hence, these results have the same effects on the interaction of the metal cations with the π-system.
AIM analysis
According to the quantum theory of atom in molecule (QTAIM), the electron density (ρ), its Laplacian (\({\nabla }^{2}\)ρ), the total electron energy density (H), and its two components (potential electron energy density, V, and kinetic electron energy density, G) in term of bond critical point (BCP) describe the chemical concepts of atom, bond, and structure. There is a prominent relation between the Laplacian and the energetic features of the critical point [53] as follows:
The electron density of the total system and the sum of the principal curvatures (\({\nabla }^{2}\)ρ = λ1 + λ2 + λ3) indicate the nature of the interaction. The perpendicular curvatures (λ1 and λ2) and the parallel curvature (λ3) of ρ at the BCP are negative and positive, respectively. The ratio of |λ1|/λ3 at the BCPs [64,65,66] measures the charge concentration. The ellipticity, ɛ, of a bond is also expressed by the two negative curvatures in a BCP as ɛ = λ1/λ2 − 1, where |λ2| <|λ1|. As can be seen in Table 3, the maximum values of the ellipticity correspond to the AC complexes in water, having the highest dielectric constant or polarity.
Two excessive situations can be defined by the Laplacian of the electronic charge density, \({\nabla }^{2}\)ρBCP. In \({\nabla }^{2}\)ρBCP > 0 and \({\nabla }^{2}\)ρBCP < 0, the electronic density is locally depleted and concentrated, respectively. Shared-shell interactions (covalent bonds) are dominated with a negative value of \({\nabla }^{2}\)ρBCP, while closed-shell interactions (i.e., ionic and van der Waals interactions) are reflected in a positive value of \({\nabla }^{2}\)ρBCP. For complexes described with \({\nabla }^{2}\)ρBCP > 0 and HBCP < 0 values, the partially covalent nature of such interaction is observed in these systems.
The calculated topological properties are summarized in Table 3. As shown in this table, all the BCPs of cation–π interaction are characterized by small values of ρBCP, \({\nabla }^{2}\)ρBCP > 0, HBCP < 0 and |λ1|/λ3 < 1. This means that the cation–π interactions are the closed-shell in nature. Since, these complexes are described with \({\nabla }^{2}\)ρBCP > 0 and HBCP < 0 values, therefore, they may indicate the partially covalent nature of such interactions.
It is prominent that the value of electron density (ρBCP) represents the strength of the cation–π interaction. For each ion group in the gas phase, the comparison of the ρBCP values with the binding energies of the complexes displays a direct relationship between them. The results obtained in this phase show the maximum value of ρBCP for the Ni2+ complex and the minimum value for the Cu+ complex. The significant changes for ρBCP are also observed in the presence of solvent. The results display that when the dielectric constant of solvent increases, the ρBCP values decrease. As can be seen in Table 3, the highest value of electron density belongs to the AC complexes in the gas phase compared to the solution phase, which can be due to the stronger interaction of the complexes in this phase. This denotes that the binding strength in the solution phase is weaker than the gas phase.
The property of cation–π interactions can also be investigated by the -G/V ratio [67, 68]: the interaction is non-covalent, when -G/V > 1, and is partially covalent, while 0.5 < -G/V < 1. The computed topological properties for AC complexes display that these interactions have a corresponding -G/V ratio in the range 0.664–0.864 (see Table 3). These results also confirm that the cation–π interaction in the considered systems is partially covalent. Figure 3 illustrates the molecular graph of a typical complex explored in this study. As shown in this figure, there are four (3, − 1) BCPs between the metal ions and the carbon atoms of the aromatic ring in this complex.

Molecular graph of AC···Mn+ complex obtained from ωB97XD/6-311++G(d,p) wave function
NBO analysis
A unique technique for assessing the delocalization effects [69] is natural bond orbital (NBO) analysis. In this work, the donor–acceptor interaction is estimated by the second-order Fock matrix. For each donor NBO (i) and acceptor NBO (j), the stabilization energy, E(2), associated with the delocalization of i → j, is calculated as follows:
According to this formula, qi provides the orbital occupancy, εi and εj correspond to diagonal elements, and F(i,j) is the off-diagonal NBO Fock matrix element [55, 70]. In the NBO analysis of these systems, the charge-transfer from the π-system of AC to the metal cations can also be evaluated as follows:
The results of the NBO analysis are reported in Table 4. As shown in this table, the most important interaction is placed between the σ-electrons of donor species and the n* of cations as acceptor groups (σ → n*cation). The results reveal that the absolute values of the obtained donor–acceptor energies, E(2), are in the ranges of 2.70–13.54 kcal mol−1. Based on the findings calculated in the solution phase, the maximum and minimum values of E(2) belong to the Mn+ and Cu+ complexes, respectively, which are directly proportional to their evaluated ∆Eion-π values. There is a good correlation between the values of E(2) versus dion-π. The correlation coefficients (R) in the gas phase, chloroform and water are 0.8620, 0.9855, and 0.9462, respectively (see Fig. 4). Inspection of the results in Table 4 also reveals that the values of E(2) in the solution phase diminish relative to the gas phase, which leads to a decrease in the charge-transfer between the components.

The correlation between the E(2) and dion-π values
The values of charge-transfer (QCT) calculated by Eq. (6) lie in the ranges of 0.004 to 0.029. As observed in Table 4, the highest and lowest QCT values are related to the Mn+ and Cu+ complexes, respectively. This quantity exhibits the significant alterations in the presence of solvent. It is obvious that with increasing the dielectric constant of solvent, the QCT values reduce. For example, the QCT value of the Fe2+ complex drops from 0.023 |e| in the gas phase to 0.021 |e| in the chloroform and to 0.019 |e| in the water. This implies that the charge-transfer in the solution is lower than the gas phase and leads to the weakening of cation–π interaction of complexes in the first case relative to latter. For each ion group (mono- or divalent) in the different media, there is a linear relationship between the QCT values and the donor–acceptor energies, E(2). The correlation between these values is given in Fig. 5. As shown in this figure, the correlation coefficients (R) for the studied complexes are equal to 0.9666 (gas phase), 0.9814 (chloroform), and 0.8536 (water).

The relationship between the E(2) and QCT values
The ΔQ values are also evaluated from the difference between the charge of free cation and the charge of cation in the complex. A look at Table 4 shows that these values are the highest for divalent complexes and the lowest for monovalent ones. It is also clear that the maximum/minimum values of ΔQ correspond to the Ni2+/Co+ complexes, respectively. In general, the results of NBO analysis show good correlations between the values of E(2), QCT and ΔQ. According to these data, the cation–π interaction of the complexes in the gas phase is stronger than the solution phase, which is due to the higher donor–acceptor energy and also the more charge transfer of the complexes in this phase. This implies that the charge-transfer properties can be a suitable parameter for estimating the strength of the cation–π interactions.
Electronic properties
An important criterion for evaluating the electronic properties of the complexes is the frontier molecular orbital (FMO) theory. The HOMO and LUMO have the nature of nucleophilic and electrophilic, respectively. The HOMO–LUMO energy gap (Eg), expressed as Eg = ELUMO − EHOMO, is a simple indicator of kinetic stability. A large energy gap is related to the higher stability and lower reactivity of a chemical system. The plot of frontier molecular orbitals (HOMO–LUMO) with their corresponding energy gap for the AC···Mn+ complex calculated by the ωB97XD method and the 6-311++G(d,p) basis set is illustrated in Fig. 6.

HOMO and LUMO plot of AC···Mn+ complex obtained at the ωB97XD/6-311++G(d,p) level of theory
Table 5 presents the quantum molecular descriptors of the AC complexes in the gas phase and solution. These quantities, which are identified as conceptual DFT parameters or chemical reactivity indicators, are defined as follows:
where μ, η, S, and ω are chemical potential [71], chemical hardness [72], global softness, and electrophilicity index [73], respectively. In an N-electron system, μ and η are characterized as the first and second derivatives of the total energy with respect to the external potential, respectively. The reciprocal of the hardness is the softness that evaluates the easiness of charge transfer and is a key factor in determining the polarizability of molecules. In addition, the electrophilic nature of a molecule is related to its electrophilicity index.
The results of Table 5 show that the values of energy gap increase on going from the gas phase to the solution (except for Ni2+ complex). This leads to increasing the stability and decreasing the reactivity of the complexes in the solution phase with respect to the gas phase. As energy gap enhances, the chemical hardness also increases. A direct relationship also exists between the chemical hardness and chemical potential values in the studied complexes. In other words, the value of chemical potential in the solution is higher than that in gas phase; since these values are negative, so all complexes are stable in the different media (see Table 5). The electronegativity (χ) [74] is defined as the negative of chemical potential, as follows: χ = − μ. Increasing the electronegativity of considered complexes in the gas phase compared to the solution phase implies that they are better electron acceptors in the first case relative to latter.
It is well known that large HOMO–LUMO gap means a hard molecule and small HOMO LUMO gap means a soft molecule. Because the soft molecules need small energy to excitation, hence, they are more polarizable than the hard molecules. Based on the results obtained in Table 5, the greater chemical softness of the complexes in the gas phase compared to the solution indicates that they are the most reactive structures in this phase (except for the Ni2+ complex). It is apparent that with decreasing the HOMO–LUMO energy gap, the softness of the complexes increases. Correlation between the softness and the energy gap is shown in Fig. 7.

Correlation between the softness (S) and the energy gap (Eg) for the AC···M complexes
The electrophilicity index (ω) is categorized according to the electrophilic nature of molecules. The weak electrophiles show ω < 0.8 eV, and the medium electrophiles demonstrate 0.8 < ω < 1.5 eV, while the strong electrophiles exhibit ω > 1.5 eV [75]. This index is a better descriptor of chemical reactivity. It is worth mentioning that the minimum energy gap is associated with the maximum electrophilicity index. As can be seen in Table 5, the ω index decreases from the gas phase to the solution. This indicates that the electrophilic nature of these complexes in the solution phase is less than the gas phase.
Molecular electrostatic potential (MPE) maps can visualize the size, shape, charge distribution, and the active sites of electronic systems in the intermolecular interactions. The electron density isosurface of the AC···Mn+ complex calculated by the ωB97XD method and the 6-311++G(d,p) basis set is illustrated in Fig. 8. The regions of attractive/repulsion potentials appear in red/blue colors, respectively. As observed in Fig. 8, the oxygen atoms of the carbonyl and hydroxyl groups (red color) with the higher electronegativity and larger electron density participate in electrophilic attacks, while the Mn+ cation and the plane of the aromatic ring (blue color) with positive electrostatic potential, participate in nucleophilic attacks.

Electron density isosurface of AC···Mn+ complex calculated by ωB97XD method and 6-311++G(d,p) basis set
Conclusions
In the present study, we have considered the interaction of AC drug with the metal cations (Mn+, Fe2+, Co+, Ni2+, and Cu+) in the gas phase and solution. The results show that the cation–π interaction strength in the gas phase is larger than that in solution. Our findings also display that the Ni2+/Mn+ complexes have the highest absolute values of ∆Eion-π in the gas/solution phases, respectively. Based on the obtained data, the complexes in water solution are the most stable. The stability order of the complexes is as follows: Fe2+ > Ni2+ > Mn+ > Co+ > Cu+. A similar trend is also observed in the chloroform. This stabilization enhances with increasing polarity of the solvent. According to the calculated HOMA values, the Cu+ complex has the greatest aromaticity and the least aromaticity is related to the Ni2+ complex. A direct relationship between the dipole moment and the polarizability of the complexes can be observed both in the gas phase and in solution. Hence, these results have the same effects on the interaction of the metal cations with the π-system.
The results of AIM analysis show that the cation–π interactions are the closed-shell in nature. Since, the studied complexes are described with \({\nabla }^{2}\)ρBCP > 0 and HBCP < 0 values, therefore, they may indicate the partially covalent nature of such interactions. Our data also indicate that the highest values of ρBCP belong to the complexes in the gas phase compared to the solution phase, which can be due to the stronger interaction of the complexes in this phase. This denotes that the binding strength in the solution phase is weaker than the gas phase. The results of NBO analysis show good correlations between the values of E(2), QCT, and ΔQ. According to these data, the cation–π interaction of the complexes in the gas phase is stronger than the solution phase, which is due to the higher donor–acceptor energy and also the more charge transfer of the complexes in this phase. It is also clear that the values of Eg and η increase on going from the gas phase to the solution (except for the Ni2+ complex). This leads to increasing the stability and decreasing the reactivity of the complexes in the solution phase with respect to the gas phase. In addition, the greater chemical softness of the complexes in the gas phase compared to the solution indicates that they are the most reactive structures in this phase.
Data availability
All data generated or analyzed during this study are included in this article.
Code availability
Not applicable.
References
Ennis ZN, Dideriksen D, Vaegter HB, Handberg G, Pottegard A (2016) Acetaminophen for chronic pain: a systematic review on efficacy. Basic Clin Pharmacol Toxicol 118(3):184–189
Warwick C (2008) Paracetamol and fever management. J R Soc Promot Health 128(6):320–323
Saragiotto BT, Abdel Shaheed C, Maher CG (2019) Paracetamol for pain in adults. BMJ 367:l6693
Klotz U (2013) Acetaminophen—a popular and widely used non-opioid analgesic. Intensiv- und Notfallbehandlung 38(2):106–110
Alchin J, Dhar A, Siddiqui K, Christo PJ (2022) Why paracetamol (acetaminophen) is a suitable first choice for treating mild to moderate acute pain in adults with liver, kidney or cardiovascular disease, gastrointestinal disorders, asthma, or who are older. Curr Med Res Opin 38:811–825
Wahba H (2004) The antipyretic effect of ibuprofen and acetaminophen in children. Pharmacotherapy 24(2):280–284
Blough ER, Wu M (2011) Acetaminophen: beyond pain and fever relieving. Front Pharmacol 2:72-1–72-6
Alves C, Borges R, Da Silva A (2006) Density functional theory study of metabolic derivatives of the oxidation of paracetamol. Int J Quantum Chem 106:2617–2623
Khuroo MS, Kamili S (2003) Aetiology and prognostic factors in acute liver failure in India. J Viral Hepat 10:224–231
Simmons DL, Wagner D, Westover K (2000) Nonsteroidal anti-inflammatory drugs, acetaminophen, cyclooxygenase 2, and fever. Clin Infect Dis 5:S211–S218
Fitzpatrick FA (2004) Cyclooxygenase enzymes: regulation and function. Curr Pharm Des 10(6):577–588
Thomas SH (1993) Paracetamol (acetaminophen) poisoning. Pharmacol Ther 60:91–120
Sanghavi BJ, Srivastava AK (2010) Simultaneous voltammetric determination of acetaminophen, aspirin and caffeine using an in-situ surfactant-modified multiwalled carbon nanotube paste electrode. Electrochim Acta 55:8638–8648
Ma JC, Dougherty DA (1997) The cation–π interaction. Chem Rev 97:1303–1324
Gokel GW, De Wall SL, Meadows ES (2000) Experimental evidence for alkali metal cation-π interactions. Eur J Org Chem 2000:2967–2978
Kim KS, Tarakeshwar P, Lee JY (2000) Molecular clusters of π-systems: theoretical studies of structures, spectra, and origin of interaction energies. Chem Rev 100:4145–4186
Cheng YH, Liu L, Fu Y, Chen R, Li XS, Guo QX (2002) Counterion effects on the cation-π interaction between alkaline earth cations and benzene. J Phys Chem A 106:11215–11220
Lehn JM (1995) Supramolecular chemistry: concepts and perspectives. 1st edn. Wiley-VCH, Weinheim, pp 199–206
Yoon J, Kim SK, Singh NJ, Kim KS (2006) Imidazolium receptors for the recognition of anions. Chem Soc Rev 35:355–360
Meyer EA, Castellano RK, Diederich F (2003) Interactions with aromatic rings in chemical and biological recognition. Angew Chem Int Ed 42:1210–1250
Lopinski GP, Wayner DDM, Wolkow RA (2000) Self-directed growth of molecular nanostructures on silicon. Nature 406:48–51
Frontera A, Quiñonero D, Garau C, Costa A, Ballester P, Deyà PM (2006) MP2 study of cation-(π)n-π interactions (n = 1–4). J Phys Chem A 110:9307–9309
Kim D, Lee EC, Kim KS, Tarakeshwar P (2007) Cation-π-anion interaction: a theoretical investigation of the role of induction energies. J Phys Chem A 111:7980–7986
Ebrahimi A, Habibi Khorassani M, Masoodi HR (2010) The role of cation–π interactions in ethylenic complexes: a theoretical NMR study. Chem Phys Lett 493:27–32
Khanmohammadi A, Raissi H, Mollania F, Hokmabadi L (2014) Molecular structure and bonding character of mono and divalent metal cations (Li+, Na+, K+, Be2+, Mg2+, and Ca2+) with substituted benzene derivatives: AIM, NBO, and NMR analyses. Struct Chem 25:1327–1342
Khanmohammadi A, Ravari F (2017) The influence of cation-π interactions on the strength and nature of intramolecular O···H hydrogen bond in orthohydroxy benzaldehyde compound. Phys Chem Res 5:57–68
Mohammadi M, Alirezapour F, Khanmohammadi A (2021) DFT calculation of the interplay effects between cation–π and intramolecular hydrogen bond interactions of 5 mesalazine drug with selected transition metal ions (Mn+, Fe2+, Co+, Ni2+, Cu+, Zn2+). Theor Chem Accounts 140:104
Mohammadi M, Mahinian M, Khanmohammadi A (2022) Theoretical study of stability and electronic characteristics in various complexes of psoralen as an anticancer drug in gas phase, water and CCl4 solutions. Chem Res Chin Univ. https://doi.org/10.1007/s40242-022-1475-522
Hassanpour A, Poor Heravi MR, Khanmohammadi A (2022) Electronic sensors for alkali and alkaline earth cations based on Fullerene-C60 and silicon doped on C60 nanocages: a computational study. J Mol Model 28:148
Pirgheibi M, Mohammadi M, Khanmohammadi A (2021) A comparative study of interplay effects between the cation-π and intramolecular hydrogen bond interactions in the various complexes of methyl salicylate with Mn+, Fe2+, Co+, Ni2+, Cu+, and Zn2+ cations. Struct Chem 32:1529–1539
Pirgheibi M, Mohammadi M, Khanmohammadi A (2021) Density functional theory study of the interplay between cation–π and intramolecular hydrogen bonding interactions in complexes involving methyl salicylate with Li+, Na+, K+, Be2+, Mg2+, Ca2+ cations. Comput Theor Chem 1198:113172
Mohammadi M, Khanmohammadi A (2019) Molecular structure, QTAIM and bonding character of cation–π interactions of mono- and divalent metal cations (Li+, Na+, K+, Be2+, Mg2+ and Ca2+) with drug of acetaminophen. Theor Chem Accounts 138:101–109
Alirezapour F, Khanmohammadi A (2021) Theoretical study on the interaction of phenylalaninal with group IA (Li+, Na+, K+) and IIA (Be2+, Mg2+, Ca2+) metal cations. J Chin Chem Soc 68:1002–1012
Alirezapour F, Khanmohammadi A (2020) The effect of cation–π interactions on the stability and electronic properties of anticancer drug Altretamine: a theoretical study. Acta Cryst C76:982–991
Mohammadi M, Khanmohammadi A (2020) Theoretical investigation on the non-covalent interactions of acetaminophen complex in different solvents: study of the enhancing effect of the cation–π interaction on the intramolecular hydrogen bond. Theor Chem Accounts 139:141
Alirezapour F, Khanmohammadi A (2020) Computational study of noncovalent interactions within the various complexes of para aminosalicylic acid and Cr2+, Mn+, Fe2+, Co+, Ni2+, Cu+, Zn2+ cations: exploration of the enhancing effect of the cation–π interaction on the intramolecular hydrogen bond. Theor Chem Accounts 139:180
Nawaz M, Hisaindee S, Graham JP, Rauf MA, Saleh N (2014) Synthesis and spectroscopic properties of pyridones—experimental and theoretical insight. J Mol Liq 193:51–59
Dutta A, Boruah B, Saikia PM, Dutta RK (2013) Stabilization of diketo tautomer of curcumin by premicellar cationic surfactants: a spectroscopic, tensiometric and TD-DFT study. J Mol Liq 187:350–358
Ai H, Bu Y, Li P, Li Z, Hu X, Chen Z (2005) Geometry and binding properties of different multiple-state glycine–Fe+/Fe2+ complexes. J Phys Org Chem 18:26–34
Banu L, Blagojevic V, Bohme DK (2012) Dissociation of deprotonated glycine complexes with Pb2+ and five transition-metal dications (Fe2+, Co2+, Ni2+, Cu2+, Zn2+): the importance of metal bond activation. Int J Mass Spectrom 330:168–173
Marino T, Toscano M, Russo N, Grand A (2006) Structural and electronic characterization of the complexes obtained by the interaction between bare and hydrated first-row transition-metal ions (Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+) and glycine. J Phys Chem B 110:24666–24673
Kurban M, Gündüz B, Göktaş F (2019) Experimental and theoretical studies of the structural, electronic and optical properties of BCzVB organic material. Optik 182:611–617
Kurban M, Gündüz B (2018) Study of structural, optical properties and electronic structure of PTCDI-C5 organic nanostructure. Chem Phys Lett 691:14–21
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su SJ, Windus TL, Dupuis M, Montgomery JA (1993) General atomic and molecular electronic structure system. J Comput Chem 14:1347–1363
Chai JD, Head-Gordon M (2008) Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys Chem Chem Phys 10:6615–6620
Krishnam R, Binkley JS, Seeger R, Pople JA (1984) Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J Chem Phys 80:3265
Iikura H, Tsuneda T, Yanai T, Hirao K (2001) A long-range correction scheme for generalized-gradient-approximation exchange functionals. J Chem Phys 115:3540
Savin A, Flad HJ (1995) Density functionals for the Yukawa electron-electron interaction. Int J Quantum Chem 56:327–332
Chai JD, Head-Gordon M (2008) Systematic optimization of long-range corrected hybrid density functionals. J Chem Phys 128:084106
Boys SF, Bernardi F (1970) The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys 19:553–566
Mennucci B (2012) Polarizable continuum model. Wiley Interdiscip Rev Comput Mol Sci 2(3):386–404
Tomasi J, Cammi R, Mennucci B, Cappelli C, Corni S (2002) Molecular properties in solution described with a continuum solvation model. Phys Chem Chem Phys 4:5697–5712
Bader RFW (1990) Atoms in molecules: a quantum theory (Reihe: International Series of Monographs on Chemistry), 1st Paperback edn. Oxford University Press, Oxford, Vol 22, pp 1–458
Biegler KF, Schonbohm J, Bayles D (2001) AIM2000: a program to analyze and visualize atoms in molecules. J Comput Chem 22:545–559
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor–acceptor viewpoint. Chem Rev 88:899–926
Krygowski TM, Cyranski MK (1996) Separation of the energetic and geometric contributions to the aromaticity of π-electron carbocyclics. Tetrahedron 52:1713–1722
Krygowski TM, Ste˛pien´ BT, (2005) Sigma- and pi-electron delocalization: focus on substituent effects. Chem Rev 105:3482–3512
Julg A, François P (1967) Recherches sur la géométrie de quelques hydrocarbures non-alternants: son influence sur les énergies de transition, une nouvelle définition de l’aromaticité. Theor Chim Acta 8:249–259
Palusiak M, Simon S, Sola M (2006) Interplay between intramolecular resonance assisted hydrogen bonding and aromaticity in o-hydroxyaryl aldehydes. J Org Chem 71:5241–5248
Güell G, Poater J, Luis JM, Mó O, Yáñez M, Sola M (2005) Aromaticity analysis of Lithium Cation/π complexes of aromatic systems. Chem Phys Chem 6:2552–2561
Lippert B (1999) Cisplatin: chemistry and biochemistry of a leading anticancer drug. Verlag Helvetica Chimica Acta, Wiley-VCH, Zürich, Weinheim, pp 1–584
Ebrahimi A, Masoodi HR, Habibi Khorassani M, Ghaleno MH (2012) The influence of cation-π and anion-π interactions on the strength and nature of N⋯H hydrogen bond. Comput Theor Chem 988:48–55
Deepa P, Kolandaivel P, Senthilkumar K (2012) Theoretical investigation of interaction between psoralen and altretamine with stacked DNA base pairs. Mater Sci Eng C 32:423–431
Bader RFW, Slee TS, Cremer D, Kraka E (1983) Description of conjugation and hyperconjugation in terms of electron distributions. J Am Chem Soc 105:5061–5068
Macchi P, Sironi A (2003) Chemical bonding in transition metal carbonyl clusters: complementary analysis of theoretical and experimental electron densities. Coord Chem Rev 238:383–412
Sola M, Mestres J, Carbo R, Duran M (1996) A comparative analysis by means of quantum molecular similarity measures of density distributions derived from conventional ab initio and density functional methods. J Chem Phys 104:636
Parra RD, Ohlssen J (2008) Cooperativity in intramolecular bifurcated hydrogen bonds: an ab initio study. J Phys Chem A 112:3492–3498
Raissi H, Khanmohammadi A, Mollania F (2013) A theoretical DFT study on the structural parameters and intramolecular hydrogen-bond strength in substituted (Z)-N(Thionitrosomethylene)-thiohydroxylamine systems. Bull Chem Soc Jpn 86:1261–1271
Weinhold F (1998) Encyclopedia of computational chemistry. In: Schleyer PVR (ed), 1st edn. Wiley, New York, pp 2699–2708
Hokmabady L, Raissi H, Khanmohammadi A (2016) Interactions of the 5-fluorouracil anticancer drug with DNA pyrimidine bases: a detailed computational approach. Struct Chem 27:487–504
Chattaraj PK, Poddar A (1999) Molecular reactivity in the ground and excited electronic states through density-dependent local and global reactivity parameters. J Phys Chem A 103:8691–8699
Pearson RG (1997) Chemical hardness-applications from molecules to solids. 1st edn. Wiley-VCH Verlag GMBH, Weinheim, pp 1–208
Parr RG, Szentpály LV, Liu S (1999) Electrophilicity index. J Am Chem Soc 121:1922–1924
Sen KD (1987) Electronegativity, structure and bonding. In: Jorgensen CK (ed), 1st edn. Springer Verlag, Berlin, Heidelberg, New York, London, Paris, Tokyo, New York, Vol 66, pp 1–205
Domingo LR, Aurell MJ, Pérez P, Contreras R (2002) Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron 58:4417–4423
Acknowledgements
The authors wish to thank Payame Noor University, Tehran, Iran, for their supports.
Author information
Authors and Affiliations
Contributions
All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
The manuscript is prepared in compliance with the Ethics in Publishing Policy as described in the Guide for Authors.
Consent to participate
The manuscript is approved by all authors for publication.
Consent for publication
The consent for publication was obtained from all participants.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Alirezapour, F., Bamdad, K., Khanmohammadi, A. et al. A computational study on acetaminophen drug complexed with Mn+, Fe2+, Co+, Ni2+, and Cu+ ions: structural analysis, electronic properties, and solvent effects. J Mol Model 28, 302 (2022). https://doi.org/10.1007/s00894-022-05305-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-022-05305-6