
Abstract
In the present investigation, we use a dual computational approach (at single molecular and solid-state levels) to explore the optoelectronic and nonlinear optical (NLO) properties of cross-shaped derivatives. The solid-state electronic band structures of the compounds 1–3 (the derivatives of tetracarboxylic acid in cross-shaped having the core of benzene (1), pyrazinoquinoxaline (2), and tetrathiafulvalene (3)) are calculated. The calculated band gaps for compounds 1–2 are found to be direct bad gaps and compound 3 to be indirect bad gap with energy gaps of 2.749, 1.765, and 0.875 eV, respectively. The important optical properties including refractive index, absorption coefficients, loss functions, and extinction coefficient of these semiconductors are calculated at bulk level to seek their potential applications as efficient optoelectronic materials. Additionally, we use the Lorentz approximation to calculate the third-order NLO susceptibilities of compounds 1–3 using the molecular hyperpolarizability and solid-state parameters. The calculated third-order NLO susceptibilities of compounds 1–3 are found to be 6.92 × 10−12, 64.0 × 10−12, and 26.3 × 10−12 esu, respectively. Thus, the present study not only provides a way to connect the calculated third-order molecular NLO polarizability to NLO susceptibilities for compounds 1–3 through Lorentz approximation but also highlights the importance of central core modifications on their NLO susceptibilities.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The recent revolution in optical devices of the twentieth century mainly depends on the rise of novel optoelectronic materials. Generally, optoelectronic materials are found as crystalline and amorphous solids. The optoelectronic materials are of great significance due to their utility in modern electronics, photonics, power devices, light emitters, and nonlinear optical applications [1, 2]. Semiconductors are used in the manufacturing of various kinds of electronic devices including diodes, transistors, and integrated circuits. Such devices have found wide use because of their efficiency, reliability, power performance, and low cost. They also found use in power systems, optical sensors, and light emitters, including solid-state lasers as discrete components [3,4,5,6,7]. Several materials exhibit good optical properties like elements of Group IVA, a combination of different families of periodic table like IIIA and VA family, some alloys and oxides, and organic molecules.
Previously, inorganic compounds like silicon, germanium, and chemical mixture were used for the development of high-quality featured optical devices. [8, 9] Over time, researchers now exploring several novel organic materials owing to their low costs, ease of fabrication, a vast variety of functional groups, and robust optical and NLO properties. For instance, M. Divya Bharathi et al. [10] worked on the third-order susceptibility of 8-hydroxyquinolinium 2-carboxy-6-nitrophthalate monohydrate owing to have 0.15 × 10−6 esu that is in-line with other species like 1.56 × 10−6 esu of 8-hydroxyquinolinium (Z)-3-carboxyacrylate(II), 2.03 × 10−6 esu of 8-hydroxyquinolinium 2-chloroacetate(I), 1.99 × 10−6 esu of 1-(carboxymethyl)-8-hydroxyquinolin-1-ium chloride, and 2.51 × 10−6 esu of 8-hydroxyquinolinium 2-chloro-5-nitrobenzoate dihydrate. Furthermore, Irfan et al. [11] investigated the optoelectronic properties of small molecules phenylimidazo[1,5-a]pyridine on bulk level. The calculations of conductivity, extinction coefficient, dielectric constant, reflectivity, and refractive indices have shown that derivatives of imidazo[1,5-a]pyridine would be efficient multifunctional organic semiconductor devices. Similarly, Aijaz et al. study the furan-based small organic semiconductor molecules (OSMs) with enhanced optoelectronic properties at the bulk level. [12] In our previous study, the first hyperpolarizability of the cross-shaped chromophores was reported using three functionals including PBEO, M06, and B3LYP. It has been found M06, the most efficient level for NLO calculation as the 2-PyQ gamma values were 257 times larger than reference para-nitroaniline (p-NA). [13]
In the light of the above literature, the cornerstone of our present investigation is to calculate the optoelectronic properties including reflectivity, conductivity, loss function, band gap structure, absorption and extinction coefficient, the density of state (DOS), dielectric function, and refractive index of three experimentally synthesized compounds that are the derivatives of tetracarboxylic acid in cross-shaped having the core of benzene (1), pyrazinoquinoxaline (2), and tetrathiafulvalene (3). Furthermore, for the first time, we use third-order molecular NLO polarizabilities to find out the bulk-level NLO susceptibilities through the Lorentz approximation. The above-entitled cross-shaped compounds are selected for investigation owing to the abundance of their π-conjugation, differences in their central cores, and similarity in terminal groups. We plan to perform a comparative analysis of their structures, optical and NLO properties which have been not reported till now.
Computational details
A dual methodological approach was used by applying the density functional theory (DFT) methods for calculation of isolated single molecular geometries and also at bulk level within periodic boundary conditions (PBC). The single molecular geometries of selected compounds (1–3) were optimized at molecular level for the calculations of hyperpolarizability using PBE0/6-311G* level of theory through Gaussian 09 suit of programs [14]. For the calculation of bulk-level electronic and optical properties including the density of states (DOS) and band structure, the Perdew-Burke-Ernzerhof (PBE) functional is used within generalized gradient approximation as implemented in Cambridge Serial Total Energy Package (CASTEP) [15,16,17] of Material Studio 7.0. [18] The CASTEP calculations within PBC were mainly based on the pseudopotential parameter, Pulay density–mixing scheme, and cutoff parameters of plane-wave basis set. Meanwhile, the six lattice parameters were selected as a = 19.44 Å, b = 30.68 Å, c = 9.11 Å, and α = β = γ = 90o for the orthorhombic compound 1. Similarly, in triclinic systems compound 2 and compound 3 were analyzed using the parameters a = 5.61 Å, b = 14.77 Å, c = 18.80 Å, α = 75.24o, β = 82.37o, and γ = 88.60o, and a = 10.02 Å, b = 16.20 Å, c = 18.33 Å, α = 76.43o, β = 87.50o, and γ = 74.08o, respectively. The sampling of Brillouin Zone (BZ) by the implementation of reciprocal space was approximated using the Monkhorst-Pack scheme. The k-point set was selected as fine mesh with (2 × 2 × 2) values. An optimized cutoff energy of 350 eV for plane-wave expansion was used with precise fast Fourier transform (FFT) [42] grid (190 × 192 × 60) (36 × 96 × 120) and (108 × 180 × 50) for compounds 1, 2, and 3, respectively. For these molecular crystals, the Broyden-Fletcher-Goldfarb-Shanno hessian modified scheme (BFGS) was being used as a minimization method, with the self-consistent field (SCF) [19]. Pseudo atomic calculation was performed for H 1s1, C 2s2, 2p2, and O 2s2, 2p4. Meanwhile, the total energy/atom convergence tolerance was 2 × 10−6 eV and Eigen-energy convergence tolerance was 1 × 10−6 eV. The other optical parameters including reflectivity R(ω), extinction coefficient k(ω), refractive index n(ω), and energy loss function L(ω) were analyzed using the following equations obtained from dielectric function (ω) based on real (€1) and imaginary (€2) parts as [20,21,22,23]:
Results and discussion
Solid-state geometries of compounds 1, 2, and 3
The geometrical structure plays an important role to modulate the optoelectronic and NLO properties of advanced functional materials. Therefore, the geometries of above-entitled compounds 1–3 and the effect of changes in geometrical structures on their optical and NLO properties are also investigated in the present study. The geometrical parameters of the selected compounds were computed and compared with the experimental crystal data at single molecular and bulk levels as shown in Fig. 1. At single molecular levels, some important bond lengths and bond angles are compared and a reasonable agreement is seen on the left side of Fig. 1 where the values in parenthesis are the experimental and taken from references [24]. While on the other hand, the chemical structures of compounds 1–3 are also energy minimized within periodic boundary conditions using PBE functional within generalized gradient approximation as implemented in CASTEP. The overviews of experimental solid-state packing and our energy minimized geometries within PBC are shown in the right side of Fig. 1. Overall similar arrangements of structures of compounds 1–3 are seen in their respective solid-state arrangements. It is reasonable to say that calculated molecular geometries are in agreement with their respective experimental structures at molecular and bulk levels.

The left side is showing single molecular optimized geometries (H-atoms are omitted) for compounds 1, 2, and 3 where bonding parameters in brackets are experimental values. The right side is showing the experimental single crystal structure packing arrangements along with their solid-state calculated geometries within PBC
Electronic band structures
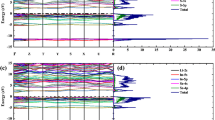
In solid-state materials, the electronic band structure is an important parameter to study the conduction of electrons from valence band to conduction band as conduction depends on the energy of the electrical band gap. The calculated band gaps of compounds 1, 2, and 3 are found to be 2.749, 1.765, and 0.875 eV, respectively. The band gap of entitled molecules was compared with each other, and a narrow band gap (0.875 eV) was found in the compound 3 that represent their enhanced ability to conduct electricity as compared to compound 1 and 2. The band gap value of compound 3 was found lesser than some ordinary semiconductors that have lower band gaps, which might show its potential for application as lower bad gap materials [25]. The calculated band structure analysis reveals that compounds 1 and 2 have direct and 3 have indirect band structures as shown in Fig. 2. The lowest energy state in the conduction band and maximum-energy state valence band are both distinguished by crystal momentum (k-vector) in the Brillouin field. It is an “indirect gap” if the k-vectors are different. When the crystal momentum of electrons and holes is the same in both the conduction band and the valence band, the band difference is considered “direct” [26, 27]. The direct band gap in compound 2 has higher energy at the Brillouin zones, i.e., F, Q, and Z in the conduction band whereas relatively low energy observed at these points in the valence band owing their least ability for the emission (luminescence) [28]. These Brillouin zones represent the reciprocal points just similar to Wigner-Seitz cells of the Bravais lattice for the elaboration of their variations. In the principle of band configuration, used in solid-state physics to evaluate the energy levels in a solid, the Fermi level may be assumed to be an electron’s potential energy level, meaning that this energy level will have a 50% chance of being filled at any given moment. A key consideration in the assessment of electrical properties is the location of the Fermi level in comparison to band energy levels. The Fermi level does not necessarily relate to a real energy level (the Fermi level exists in the band gap in an insulator) nor does it allow band structure to occur [29, 30].

The electronic band structures of the compound 1 (on the top), compound 2 (at the middle), and compound 3 (at the bottom) with the zoomed diagram (− 0.1 to 0 eV) on the right of each
Density of states
To estimate the contributions of individual orbitals (s and p orbitals) towards the total absorption, total density of state (TDOS) and partial density of state (PDOS) analyses were performed for molecules 1–3. The TDOS study expressed the total number of states present within the molecule. It can be seen from Fig. 3 that compound 1 has the highest states near 2 eV that correspond to their band gap energy. Furthermore, s subshell involvement is less as compared to p subshell while there is an intense peak observed at − 5 eV in which the role of p subshell is maximum. In compound 2, there is an overall less amplitude of TDOS as compared to other compounds in the study. The maximum TDOS is found to be about 30 electrons/eV at − 5 eV in compound 2. After careful analysis, it is observed that compound 1 has maximum contributions in TDOS from s and p orbitals that are higher than the other two compounds. It is clear from Fig. 3 that the p orbital played a major role in these compounds for the overall TDOS. The s-orbitals are representing the energy bands in the deeper valence band (VB) from − 3 to − 2 eV in compound 1 with no participation in compounds 2 and 3. The p orbitals are dominating the TDOS as well as the PDOS in VB as well as in conduction band (CB). The TDOS summary displays the dominance of p orbitals near the Fermi level of compound 1.

The partial and total density of state (PDOS and TDOS) of compounds 1, 2, and 3 from left to right, respectively
Dielectric function
The entitled molecules first tested on 001, 100, 010 vectors but it is found that there are no major differences in their optical properties like dielectric function. That is why all the calculations performed using polycrystalline. Dielectric function is an important factor to describe the properties of the compound as it plays a chief role in the applications of charge screening, capacitance, [31] and electrical charge storage. [32] It has two parts, i.e., the real and imaginary parts that describe the degree of polarization (dielectric constant) and dielectric losses, respectively. [33] It was observed that compound 1 has a maximum real dielectric value of 1.7 at the wavelength of about 475 nm while the imaginary part has a maximum value (1.1) at about 400 nm and then it becomes steady throughout the graph. However, the compound 2 showed unusual variation in the real and imaginary parts of dielectric functions in such a way that real part value falls up to 200 nm from 1.0 to 0.5 exhibiting their relaxation phenomenon and then exponentially increases till 700 nm up to 2.5 value of the dielectric function as molecules are unable to relax at high frequency due to low dielectric constant value. Similarly, the compound 3 has a non-uniform trend of variation in the beginning as real part varies from 1.0 to 0.5 at the wavelength of about 50 to 200 nm and then rapidly increases to 2.0 as the wavelength increases and remains uniform after wavelength of 400 nm whereas the imaginary part has maximum dielectric function value (1.25) at about 350 nm that has been represented in Fig. 4.

The dielectric function of compound 1 (on the left side), compound 2 (at the middle), and compound 3 (the on right side)
Absorption and conductivity
In chemistry, absorption of radiation is associated with the excitation of atoms, particles, or charge species from the ground state to an excited state and also responsible for the conduction of electricity. [34] Hence, the absorption spectra of each molecule were calculated in which compound 2 has the maximum absorption at about 250 nm while the compound 1 and compound 3 have less intense absorptions near 300 nm. As the absorption significantly occurs in the visible region (see Fig. 5), so the collection of solar radiation for solar cell applications is significant from these compounds. These compounds can also play a significant role in the image sensor of the ordinary visible camera and ultraviolet security camera as absorption of these molecules also falls under the UV-Visible regions. These molecules can also be used as UV sensors as well as to filter the infrared radiations in the fields of aerospace, military, biological, industrial, and communication. [35]

The UV-Vis spectra of compounds 1–3 (left to right) where the visible region is shown by color bars
When the conductivities of these molecules were calculated and compared, it was found that no molecule starts conduction before 40 nm owing to that there is electronic delocalization at a lower wavelength. According to Fig. 6, the real and imaginary parts of conductivity for these three compounds have almost equal magnitude up to the range of 150–200 nm and then imaginary and real conductivities increase oppositely at 200–225 nm that are related to their dielectric functions. The values of the imaginary part fall representing the enhancement of capacitance property while the real part of conductivity increase that represents the electrical volume in compound 1. In the case of compound 2, there are two sharp peaks observed at 100 nm for the real and imaginary conductivities of about 0.4 and − 0.01 fs−1, respectively. Then afterward, imaginary and real conductivities increase up to 200 nm and then real conductivity gradually decreases while imaginary conductivity falls exponentially indicating their ability to store electrical charge. A similar pattern was observed in compound 3, where a small change occurs in both conductivities up to 200 nm while the maximum conductivity of about 0.58 fs−1 observed at 350 nm. Interestingly, these compounds have uniform conductivities including both real and imaginary parts in the visible range.

The real and imaginary conductivities of compounds 1, 2, and 3 (from left to right)
Refractive index (n) and extinction coefficient (k)
Refractive index (n) describes the movement of electromagnetic radiation when it passes through the medium and changes occur as incident light reflected from the medium indicating their thickness that is illustrated in Fig. 7. The refractive index is inversely related to the thickness of the material in such a way that an increase in the number of layers results in the lowering of the refractive index and extinction coefficient. [36] It was observed that refractive index variation starts from 50 nm at about 1.0 that gradually decreases up to 275 nm then increases to 450 nm at about 1.45 and remains steady up to 1000 nm indicating uniform geometry in compound 1. Similarly, in compound 2, there is a sharp peak of extinction coefficient and refractive index while the refractive index increases to 1.7 in the range of 600–1000 nm. Meanwhile, the extinction coefficient decreases exponentially up to 0.1 at about 1000 nm. Interestingly, compound 3 has intermediate values of refractive index and extinction coefficient and come very close to each other at about 0.6 in the range of 200 nm and then move smoothly on the visible region with n near 1.5 and k at about 0.15.

Refractive index (n) and extinction coefficient (k) of compounds 1, 2, and 3
Reflectivity and loss function
The reflectivity of a material is related to its absorption coefficient. When the reflectivity of our selected molecules was compared with each other, it was observed that compound 1 has maximum reflectivity (0.05) in the range of 250–400 nm indicating the width of the compound while compound 2 exhibits an intense peak of reflectivity at 200 nm and there is a broad peak observed in compound 3 starting from 200 nm and maximum at about 300 nm, corresponding to their absorption coefficient as described above. [37] The collected results are shown in Fig. 8.

The reflectivity of compound 1 (on the left side), compound 2 (at the middle), and compound 3 (on the right side)
Similarly, when the loss function of these compounds was compared, it was found that their peaks are much intense and sharper than reflectivity. Compound 1 exhibits unavailing loss function at about 0.8 representing excitations having a maximum range near 300 nm. The compound 2 showed extraordinarily higher value of loss function nearly 1.6 that is found at sharp 200 nm. Meanwhile, compound 3 has a broad range of loss function (1.0) that is higher than the compound 1 as there is more area under the curve that correspond to their excitations of electrons in the ultraviolet region [38] that is shown in Fig. 9.

The loss function of compounds 1–3 (from left to right)
Third-order NLO polarizability and susceptibility
Nonlinear optical materials are playing a crucial role in the modern hi-tech era. Their applications range from laser frequency modulation to digital data writing and telecommunications, etc. Organic compounds are considered novel candidates for the linear and nonlinear optical (NLO) microscopic polarizability at the molecular level and macroscopic susceptibility at bulk levels. [39, 40] These organic compounds have unique features like donor-π-accepter groups and substantial response in light-matter interactions. [41] The above discussion reveals that entitled molecules possess good optoelectronic properties so we have calculated NLO properties of compounds 1–3. Unlike the several previous NLO studies, which reported only NLO polarizabilities of organic molecules, herein, we have also calculated the third-order NLO susceptibilities (χ3) at bulk using Lorentz approximation. The third-order NLO susceptibility corresponds to linear refractive index as given in the following Eq. 6.
where χ3 is the third-order NLO susceptibility, N is the number density of molecules, nr is the linear refractive index, while γ is the molecular third-order NLO polarizability of the compounds 1–3 as calculated using PBE0/6-311G* level of theory. The calculated results of compounds 1–3 are given in Table 1 along with nonlinear optical properties of p-NA, which is a prototype organic molecule, often used as a reference in comparison to newly designed organic compounds.
To analyze the reliability of these systems, χ3 values of the entitled systems are also compared with the fullerene (C60: χ3=7 × 10−12 esu) [42], 8HQNP (χ3=35.08 × 10−12 esu) [10], DBA (χ3= 0.01 × 10−12 esu) [43], and Me-ANC (χ3=0.08 × 10−12 esu) [44]. The χ3 values of the selected molecules are arranged as DBA < Me-ANC < p-NA < 1 < C60 < 3 < 8HQNP < 2. There is a direct relationship among the third-order NLO polarizabilities (γ), refractive indices (n), and third-order NLO susceptibilities (χ3). After careful analysis of Table 1, it is observed that compound 2 showed the highest values of χ3 (64.0 × 10−12 esu) and γ (663.4 × 10−36 esu) as compared to compounds 1, 3, p-NA, Me-ANC, 8HQNP, and DBA. The calculated values of third-order NLO susceptibilities at bulk are 162, 2324, and 896 times higher as compared to the χ3 the amplitude of p-NA for the compounds 1, 2, and 3, respectively. Therefore, it is concluded that if the compounds have higher values of γ and n, there will be a surge in the value of χ3. Hence, this comparison suggests that these compounds can be used as efficient materials in the field of nonlinear optics (NLO) and other optical applications.
Conclusions
In the present investigation, the quantum chemical study for compounds 1, 2, and 3 have been performed to evaluate their potential as efficient optoelectronic and NLO materials. The molecular third-order NLO polarizabilities were found to be 201.2 × 10−36, 663.4 × 10−36, and 444.4× 10−36 esu for compounds 1–3, respectively, at PBE0/6-311G* level of theory. The calculated molecular third-order NLO polarizabilities were used along with bulk-level solid-state parameters to approximate their third-order NLO susceptibilities (a bulk-level property). The bulk NLO susceptibilities were calculated to be 6.92 × 10−12, 64.0 × 10−12, and 26.30 × 10−12 esu for compounds 1–3, respectively. It is also found that compound 2 has maximum absorption with a sharp peak in non-vacuum UV region at about 250 nm as compared to the compounds 1 and 3, which have peaks near-visible region ranges from 300 to 450 nm. For compound 2, its dielectric function exhibited higher value in the real and imaginary parts (unlike compounds 1 and 3) where real part height values range from 1.0 to 0.5 at 200 nm while 2.5 amplitude was observed at 700 nm. For compound 2, its refractive index was found to be 1.7 in the range of 600–1000 nm. The band gap of compound 2 has an intermediate energy of 1.765 eV as compared to compounds 1 and 3 that can be used for semiconducting properties. From the above discussion, it is concluded that compound 2 possesses much better optoelectronic and NLO properties as compared to compounds 1 and 3, and some reference molecules as discussed. In short, the entitled compounds are useful and these can play a key role for efficient optical and NLO properties both in single molecular state and in solid-state as well.
References
Lin Q, Armin A, Nagiri RCR, Burn PL, Meredith P (2015) Electro-optics of perovskite solar cells. Nat Photonics 9:106
Forrest SR (2000) Active optoelectronics using thin-film organic semiconductors. IEEE J Sel Top Quantum Electron 6:1072–1083
Mhibik O, Forget S, Ott D, Venus G, Divliansky I, Glebov L et al (2016) An ultra-narrow linewidth solution-processed organic laser. Light Sci Appl 5:e16026
Brabec CJ (2004) Organic photovoltaics: technology and market. Sol Energy Mater Sol Cells 83:273–292
Hasegawa H, Sato T (2005) Electrochemical processes for formation, processing and gate control of III–V semiconductor nanostructures. Electrochim Acta 50:3015–3027
Ruiz C, García-Frutos EM, Hennrich G, Gómez-Lor B (2012) Organic semiconductors toward electronic devices: high mobility and easy processability. J Phys Chem Lett 3:1428–1436
Cheng C, Zhu C, Huang B, Zhang H, Zhang H, Chen R et al (2019) Processing halide perovskite materials with semiconductor technology. Adv Mater Technol 4:1800729
Steigerwald ML, Brus LE (1990) Semiconductor crystallites: a class of large molecules. Acc Chem Res 23:183–188
Jackson TN, Lin Y-Y, Gundlach DJ, Klauk H (1998) Organic thin-film transistors for organic light-emitting flat-panel display backplanes. IEEE J Sel Top Quantum Electron 4:100–104
Bharathi MD, Bhuvaneswari R, Srividya J, Vinitha G, Prithiviraajan R, Anbalagan G (2018) Synthesis, optical, experimental and theoretical investigation of third order nonlinear optical properties of 8-hydroxyquinolinium 2-carboxy-6-nitrophthalate monohydrate single crystal. J Phys Chem Solids 113:50–60
Irfan A, Chaudhry AR, Al-Sehemi AG, Assiri MA, Hussain A (2019) Charge carrier and optoelectronic properties of phenylimidazo [1, 5-a] pyridine-containing small molecules at molecular and solid-state bulk scales. Comput Mater Sci 170:109179
Chaudhry AR, Haq BU, Muhammad S, Laref A, Irfan A, Algarni H (2019) Structural, electronic and optical properties of furan based materials at bulk level for photovoltaic applications: a first-principles study. Comput Theor Chem 1147:20–28
Muhammad S, Chaudhry AR, Al-Sehemi AG (2017) A comparative analysis of the optical and nonlinear optical properties of cross-shaped chromophores: quantum chemical approach. Optik. 147:439–445
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR et al Gaussian 09; Gaussian, Inc: Wallingford, CT, 2009. Gaussian 09; Gaussian, Inc: Wallingford, CT, 2009
Segall M, Lindan PJ, Probert MA, Pickard CJ, Hasnip PJ, Clark S et al (2002) First-principles simulation: ideas, illustrations and the CASTEP code. J Phys Condens Matter 14:2717
Cardona M, Pollak F (1971) Optical properties and model density of states. The physics of opto-electronic materials. Springer 81–112. https://doi.org/10.1007/978-1-4684-1947-4_4
Volovik G (1993) Superconductivity with lines of gap nodes: density of states in the vortex. Inst. Teoreticheskoj Fiziki 58(5–6):457–461
Clark SJ, Segall MD, Pickard CJ, Hasnip PJ, Probert MI, Refson K et al (2005) First principles methods using CASTEP. Z Kristallogr Cryst Mater 220:567–570
Press WH, Vetterling W, Teukolsky SA, Flannery BP, Greenwell Yanik E (1994) Numerical recipes in fortran--the art of scientific computing. SIAM Rev 36:149
Bhattacharya B, Sarkar U (2016) The effect of boron and nitrogen doping in electronic, magnetic, and optical properties of graphyne. J Phys Chem C 120:26793–26806
Bhattacharya B, Paul D, Sarkar U (2019) Electronic and optical properties of XN-ynes (X= B, Al, Ga): a first-principle study with many-body effects. Appl Surf Sci 495:143612
Samantaray C, Sim H, Hwang H (2005) The electronic structures and optical properties of BaTiO3 and SrTiO3 using first-principles calculations. Microelectron J 36:725–728
Okoye C (2003) Optical properties of the antiperovskite superconductor MgCNi3. J Phys Condens Matter 15:833
Hisaki I, Affendy NE, Tohnai N (2017) Precise elucidations of stacking manners of hydrogen-bonded two-dimensional organic frameworks composed of X-shaped π-conjugated systems. CrystEngComm. 19:4892–4898
De Vos A (1980) Detailed balance limit of the efficiency of tandem solar cells. J Phys D Appl Phys 13:839
Ünlü H (1992) A thermodynamic model for determining pressure and temperature effects on the bandgap energies and other properties of some semiconductors. Solid State Electron 35:1343–1352
So FF, Forrest SR (1989) Organic-on-inorganic semiconductor photodetector. IEEE Trans Electron Devices 36:66–69
Acharyya P, Pal P, Samanta PK, Sarkar A, Pati SK, Biswas K (2019) Single pot synthesis of indirect band gap 2D CsPb 2 Br 5 nanosheets from direct band gap 3D CsPbBr 3 nanocrystals and the origin of their luminescence properties. Nanoscale. 11:4001–4007
Paul D, Deb J, Bhattacharya B, Sarkar U (2018) Electronic and optical properties of C 24, C 12 X 6 Y 6, and X 12 Y 12 (X= B, Al and Y= N, P). J Mol Model 24:204
Bhattacharya B, Deb J, Sarkar U (2019) Boron-phosphorous doped graphyne: a near-infrared light absorber. AIP Adv 9:095031
Bessler R, Duerig U, Koren E (2019) The dielectric constant of a bilayer graphene interface. Nanoscale Adv 1:1702–1706
Dimitrakopoulos C, Purushothaman S, Kymissis J, Callegari A, Shaw J (1999) Low-voltage organic transistors on plastic comprising high-dielectric constant gate insulators. Science. 283:822–824
Iqbal J, Yahia I, Zahran H, AlFaify S, AlBassam A, El-Naggar A (2016) Linear and non-linear optics of nano-scale 2′, 7′ dichloro-fluorescein/FTO optical system: Bandgap and dielectric analysis. Opt Mater 62:527–533
Bakulin AA, Rao A, Pavelyev VG, van Loosdrecht PH, Pshenichnikov MS, Niedzialek D et al (2012) The role of driving energy and delocalized states for charge separation in organic semiconductors. Science. 335:1340–1344
Yang D, Ma D (2019) Development of organic semiconductor photodetectors: from mechanism to applications. Adv Opt Mater 7:1800522
Lehmuskero A, Kuittinen M, Vahimaa P (2007) Refractive index and extinction coefficient dependence of thin Al and Ir films on deposition technique and thickness. Opt Express 15:10744–10752
Cardona M (1961) Fundamental reflectivity spectrum of semiconductors with zinc-blende structure. J Appl Phys 32:2151–2155
Knupfer M, Fink J, Fichou D (2001) Strongly confined polaron excitations in charged organic semiconductors. Phys Rev B 63:165203
Brik M (2010) First-principles calculations of electronic, optical and elastic properties of ZnAl2S4 and ZnGa2O4. J Phys Chem Solids 71:1435–1442
Deb J, Paul D, Sarkar U (2020) Density functional theory investigation of nonlinear optical properties of T-graphene quantum dots. J Phys Chem A 124:1312–1320
Kubodera K, Kobayashi H (1990) Determination of third-order nonlinear optical susceptibilities for organic materials by third-harmonic generation. Mol Cryst Liq Cryst 182:103–113
Kafafi Z, Lindle J, Pong R, Bartoli F, Lingg L, Milliken J (1992) Off-resonant nonlinear optical properties of C60 studied by degenerate four-wave mixing. Chem Phys Lett 188:492–496
Kiran AJ, Chandrasekharan K, Nooji SR, Shashikala H, Umesh G, Kalluraya B (2006) χ (3) measurements and optical limiting in dibenzylideneacetone and its derivatives. Chem Phys 324:699–704
Mathew E, Salian VV, Joe IH, Narayana B (2019) Third-order nonlinear optical studies of two novel chalcone derivatives using Z-scan technique and DFT method. Opt Laser Technol 120:105697
Muhammad S, Shehzad RA, Iqbal J, Al-Sehemi AG, Saravanabhavan M, Khalid M (2019) Benchmark study of the linear and nonlinear optical polarizabilities in proto-type NLO molecule of para-nitroaniline. J Theor Comput Chem 1950030
Acknowledgments
The authors from the King Khalid University extend their appreciations to Deanship of Scientific Research at King Khalid University for funding the work through the general research project (RGP2-109-41).
Funding
Shabbir Muhammad, Abdullah G. Al-Sehemi, and Zouhaier Aloui acknowledge the funding from Deanship of Scientific Research at King Khalid University, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
Rao Aqil Shehzad and Shabbir Muhammad conceived the research idea while Javed Iqbal and Abdullah G. Al-Sehemi helped in performing calculations. Muhammad Yaseen, Zouhaier Aloui, and Muhammad Khalid have contributed in interpretations, plotting, and writing of results, respectively.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
No animal/human data is used here.
Consent for publication and participation
The consent of all authors and institutes is obtained for publication of present work.
Data transparency
All authors acknowledge that all data of present paper as well as software applications and code availability support their published claims and comply with field standards.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 23 kb)
Rights and permissions
About this article

Cite this article
Shehzad, R.A., Muhammad, S., Iqbal, J. et al. Exploring the optoelectronic and third-order nonlinear optical susceptibility of cross-shaped molecules: insights from molecule to material level. J Mol Model 27, 12 (2021). https://doi.org/10.1007/s00894-020-04619-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-020-04619-7