
Abstract
In this paper, we present the quantum electronic study of iso-octane, based on MP2 and B3LYP methods using the 6-311++G(d,p) basis set. In addition to conformational stability and internal rotation barriers studies, the delocalization energies associated with the internal charge transfer (ICT) within each of the six lowest energy conformers were evaluated using NBO analysis. With the aim to differentiate even more between these conformers, the energy gap between HOMO and LUMO orbitals, chemical softness, and first-order hyperpolarizability (nonlinear optics property) were evaluated. Similarly, their spectral behavior was investigated at different levels; the ultraviolet (UV) absorption bands were assigned using molecular orbitals data obtained by TD-B3LYP calculations with 6-311++G(d,p) basis set, while carbon 13C NMR and proton 1H signal peaks were assigned using the GIAO-B3LYP/6-311++G(d,p) method. In addition, the normal mode calculations of the most and least stable conformers using a scaled force field in terms of nonredundant local symmetry coordinates were carried out to approach the vibrational spectra temperature dependency.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The branched hydrocarbon 2,2,4-trimethylpentane (TMP), often referred to iso-octane, commonly results in the processing of crude petrol and therefore is of great interest in the petrochemical industry [1]. Moreover, turbocharging causes excessive detonation, accompanied by an extreme rise of temperature and pressure in the combustion chamber leading to the knocking noise [2]. Because the latter is very damaging for both human and equipment, the use of gasolines with an antiknock value, i.e., the ability to withstand the compression, has proven vital. This ability is measured by the octane number as an indicator of energy efficiency. In this regard, iso-octane is considered as a reference (index 100) in terms of knock resistance against heptane (index 0) [3]. In addition, the oxidation of hydrocarbons forms the basis of most processes for producing polymers (ethylene oxide and propylene oxide, maleic and terephthalic acids, etc.). For instance, iso-octane is the recurring structural subunit of polypropylene and of many polypropionate derived natural products [4,5,6,7]. However, the high temperature kinetic mechanism of the oxidation in its propagation and termination phases requires a beforehand initiation phase where a hydrogen is abstracted from the hydrocarbon, before oxidizing its radical form. This resulted in the idea to study the conformational behavior of the trimethyl-pentanes, and more particularly iso-octane, which has multiple steric environment hydrogens. Through this study, we explored the structural and electronic properties, determined the optimal conformers and the rotational barriers impeding movement between them, and elucidated the impact of the kinetic and interactional components of the molecule on the vibrational spectra (IR/Raman) in liquid phase, using scale factors of local symmetry force constants [8]. The fundamental vibrational modes were performed with their potential energy distributions (PED-s) and attributed to corresponding observed frequencies for the six lowest energy conformers. Nonlinear activity, particularly first hyperpolarizability, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), molecular electrostatic potential (MEP) [9], natural bond orbital (NBO) analyses [10], and NMR and UV spectra [11, 12] predictions have been also investigated, in order to have not only an insight into electronic properties of this molecule but also to differentiate between its conformers.
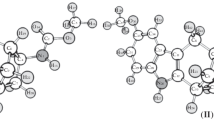
The carbon positions for each conformer are well known by evaluating the two central C–C bonds τ1 = C1–C2–C3–C4 and τ2 = C2–C3–C4–C5. The torsional angle τ1 is taken to be positive if, when looking from C2 along the C2–C3 bond, C4 is in the clockwise sense with respect to C1. The initial configuration and backbone torsional angles, as well as the numbering of the carbon atoms, are shown in Fig. 1 [13].

Backbone torsional angles and the carbon atoms numbering for 2,2,4-trimethylpentane (iso-octane)
Theoretical characterizations
The set of programs Molpro was used to carry out the ab initio and DFT optimization [14, 15]. Full geometry optimizations were performed using the method of complete relaxation without any symmetry constraint. Standard gradient techniques at Møller–Plesset perturbation method level MP2 and DFT\B3LYP level, which uses the Becke’s hybrid exchange functional B3 and the Lee–Yang–Parr nonlocal correlation functional, were calculated using the 6-311++G(d,p) basis set [16,17,18].
Before its relaxed optimization, the total electron energy surface was constructed in steps of 30°, combining the values of τ1 ranging from 0 to 120° considering the C3v local symmetry of the tert-butyl group and the values of τ2 ranging from 0 to 360° (Fig. 1). It must be emphasized that the axis of rotation of the tert-butyl group coincides with the central C2C3 bond, while the axis of rotation of the isopropyl group (C2v local symmetry) does not [19]. Only the most stable conformer and the five secondary ones obtained by the optimization calculations are presented in Newman form to give a clear description of the interactions between close groups for each conformer. The transition from one conformer to another needs to overcome the rotational barrier due to the torsion of tert-butyl or isopropyl group, calculated by fixing only the dihedral τ1 or τ2 respectively and allowing the variation of all the other parameters. All structures were visualized by employing the Chemcraft 1.8 program [20].
After complete optimization, the vibrational frequencies for these structures were computed from the analytical second derivatives of energy (Hess matrix), at the same levels of methods, with the Gaussian 09 program [21], in order to confirm that they are the true minima on one hand and to carry out their vibrational analysis on the other hand. The unscaled vibrational frequencies are larger than the experimental values because of basis set incompleteness, negligence of electron correlation, and vibrational anharmonicity. Therefore, for reasonable frequency matching, we scaled the ab initio quadratic force constant matrix according to the hessian matrix with the possibility of refinement of the scale factors. Indeed, it is necessary to modulate the quantum force constants by scaling factors C, which are determined from the least-squares adjustment of the calculated frequencies on the experimental ones [22].
To help assign vibrational modes, potential energy distributions (PED-s) have been computed using the Gar2ped program [23].
On the basis of the Raman scattering theory, the Raman intensities were also predicted with Gaussian 09, using the following relationship:
where Si are the Raman activities, ν0 is the exciting frequency in cm−1, νi is the vibrational wave number of the ith normal mode, h, c, and kb are the fundamental constants and f is a suitably chosen common normalization factor for all the peak intensities [24, 25].
Natural bonding orbital calculations were performed at B3LYP/6-311++G(d,p) using pop = nbo code as implemented in the Gaussian 09 package. The interactions due to the overlap between bonding and antibonding orbitals give rise to intramolecular charge transfer (ICT), causing stabilization of the molecule [26]. These interactions are observed as an increase in electron density in antibonding orbital that weakens the respective bond. The delocalization energies associated with the ICT were examined using the Second Order Perturbation Theory analysis of the Fock matrix in the NBO method [27]. For each donor (i) and acceptor (j), the stabilization energy E(2) associated with the delocalization i/j is calculated as:
where qi is the orbital occupancy, Ei and Ej are the diagonal elements and F(i,j) is the off-diagonal NBO Fock matrix element [28].
The calculations of polarizability α and first-order hyperpolarizibility β were performed on the optimized geometry to understand the nonlinear optical (NLO) behavior of iso-octane conformers [29]. Their electronic energy sensitivity to an external electric field Fi is expressed as follows:
where Eo is the electronic energy of the unperturbed molecule, Fi is the external field at the origin, μi, αij, and βijk are the components of dipole moment, polarizability, and first-order hyperpolarizability, respectively. The total static dipole moment μ, the mean polarizability α0, the anisotropy of the polarizability Δα, and the mean first-order hyperpolarizability β0, using the x, y, and z components are defined as [30,31,32]:
Besides this work, we predicted 13C and 1H NMR chemical shifts. It is well known that the inductive attractor effect (-I), attracting and distancing electrons from their nucleus, decreases electron density and electronic diamagnetism near the nucleus. To this diamagnetism, a screen constant σi is associated. It reflects the degree of shielding of the nucleus with regard to its environment, thus giving rise to a peak of resonance, in accordance with NMR fundamental formula.
where γ is the gyromagnetic ratio and B0 is the magnetic field intensity, and the chemical shift, which is the resonant frequency of a nucleus relative to the tetramethylsilane (TMS) standard in a magnetic field, is given by:
Therefore, the lower the screen constant (deshielding effect), the larger the chemical shift δ and the lower the field of NMR signal.
It should also be noted that, in addition to the negative sensitivity of the nucleus chemical shift to its surrounding electron density and thus to the electronegativity of the neighboring atoms, it is favorably sensitive to magnetic anisotropy resulting from the electronic current associated with the delocalization of electrons.
For iso-octane, we calculated the chemical shift of both 13C and 1H for the six A–F conformers, in order to differentiate them, once more, and to verify the compatibility of their values to the experiment. The structure of the six conformers was optimized at the B3LYP/6-311+G(d,p) level. Then, the gauge-including/invariant atomic orbital (GIAO) [33] 1H and 13C chemical shift calculations are performed by the same basis set in CDCl3 solution by IEFPCM model [34].
On the other hand, because DFT and time dependent DFT (TD-DFT) can offer a highly acceptable prediction of the electronic and optical properties, the excitation energies of the title molecule were also performed at TD-B3LYP/6-311++G(d,p), in the gas phase and in DMSO solvent, to reproduce the UV spectrum [35, 36].
Results and discussion
Geometry optimization and conformational stability
Taking into account the symmetry of the tert-butyl group and the coincidence of its local symmetry axis with the rotational bond C2C3 contrary to the isopropyl group, the exploration of the iso-octane conformational space generated 48 conformations combining τ1 and τ2 values as presented in Fig. 2.

Distribution of the 2,2,4-trimethylpentane conformers with respect to their initial torsion angles τ1 and τ2 by B3LYP/6-311++G(d,p) method
Ab initio MP2/6-311++G(d,p) and B3LYP/6-311++G(d,p) levels provide two nearly equivalent minima A and B in terms of interactions, two first secondary conformers C and D in a range of 0.6 kcal mol–1, and two nearly equivalent high secondary conformers in a range of 3.6 kcal mol–1, the conformers C and D are equivalent by effect of symmetry with respect to the trans main chain, whereas A and B or E and F are equivalent in terms of interactions between atoms only. In addition to torsional angles differences between conformers, the conformational variability also concerns the CCC angles. It reaches a maximum value (6°) for the central angle C2C3C4 for both methods.
By comparing B3LYP/6-311++G(d,p) and MP2/6-311++G(d,p), it is noteworthy that the former is slightly more stabilizing as the energy is decreased about 655 kcal mol–1, while the latter renders values of C2C3, C3C4 bond lengths and C2C3C4 angle, for the six conformers A–F, shorter by about 0.01 Å and 1° than the B3LYP ones, while almost all the other bond lengths and angles remain unchanged for the two methods. Regarding the dihedral angles, we noticed that the variation between angles does not exceed 4° from one method to another, especially for C and D conformers (Table 1, Fig. 3). Additionally, the computed bond lengths and bond angles of the titled compound were compared with their experimental data [37]. The MP2/6-311++G(d,p) values obtained for the angle α (C1–C2–C3) and distance r (C1–C2) are closer to the experiment compared to the B3LYP/6-311++G(d,p) values (Table 1).

Newman projections of the six lowest energy conformers of 2,2,4-trimethylpentane
In the light of the optimization results, the syn-pentane interaction, which appears between every fifth carbon atom, is considered the major factor influencing the stability of conformers. This explains the weakness of strain caused by tert-butyl/isopropyl interaction in A and B conformers compared to the secondary ones (Fig. 3).
Isopropyl barriers to internal rotation
The rotational barriers between the most stable conformers A and B or between secondary conformers C and E or D and F and their correspondent inversion barriers were evaluated at both MP2 and B3LYP using 6-311++G(d,p) basis set. All these transitions A (τ1 = 55, τ2 = 140) → B (τ1 = 66, τ2 = 96), C (τ1 = 42, τ2 = 63) → E (τ1 = 74, τ2 = −54), and D (τ1 = 78, τ2 = 170) → F (τ1 = 48, τ2 = −76) have the particularity of being one-dimensional rotation around C3C4 bond, knowing that the torsion angle τ2 was kept at constant value, in 5° intervals, while all the other angles and bond lengths were optimized. Moreover, with τ1 near 60, the evolution of the rotational transition throughout the total interval of τ2 [−180, 170] enabled us to identify the equivalence between C → E and D → F rotational transitions, due to a Cs symmetry of the molecule for τ2 = 120.
According to the results collected in Table 2 and presented in Figs. 4 and 5, the MP2/6-311++G(d,p) or B3LYP /6-311++G(d,p) rotational barrier of A → B transition and its inversion barrier are very weak compared to their corresponding C → E or D → F transitions. By comparing the two methods, MP2/6-311++G(d,p) C → E or D → F rotational barriers and their corresponding inversion barriers were found to be larger by about 1 kcal mol–1 than B3LYP/6-311++G(d,p) values. Obviously, the values of C → E or D → F rotational barriers and their inversion barriers are high because they have to overcome the strong interaction of the tert-butyl and isopropyl groups.

Evolution of B3LYP/6-311++G(d,p) rotational barrier to torsional angle τ2 = C2C3C4C5 for 2,2,4-trimethylpentane

B3LYP/6-311G++(d,p) and MP2/6-311G++(d,p) rotational barriers to torsional angle τ2 = C2C3C4C5 for 2,2,4-trimethylpentane
Considering the absence of experimental rotational barriers for iso-octane and the abundance of experimental data for iso-butane (methylpropane), we assessed the consistency of the methods MP2/6-311++G(d,p) and B3LYP/6-311++G(d,p) by evaluating the internal rotation barrier of methyl with respect to the only existing stable conformation of iso-butane (Fig. 1S in Supplementary information). We thus found that the 3.56 kcal mol–1 value of the MP2/6-311++G(d,p) rotation barrier is closer to the experiment than 3.24 kcal mol–1, calculated by B3LYP/6-311++G(d,p), since its evaluation from microwave spectra [38] and from two thermodynamic results [39] gives respectively 3.90, 3.62, and 3.87 kcal mol–1. However, apart from the calculation of the rotation barrier, which is part of the kinetic study, the B3LYP/6-311++G(d,p) is more stabilizing in terms of energy and thus thermodynamically and spectroscopically. This is why the DFT prediction of electronic and spectroscopic properties was chosen.
HOMO–LUMO and molecular electrostatic potential (MEP) analyses
The energy gap between HOMO and LUMO orbitals is directly related to chemical softness and the chemical reactivity of a molecule in the frontier molecular orbitals (FMOs) theory [40]. The huge gap explains the high stability and low reactivity, as is the case, for instance, with the saturated hydrocarbons and their oxidation processes. The values of HOMO, LUMO, energy gap, and softness for the six conformers are collected in Table 3. The pictorial diagram of HOMO and LUMO energy levels is presented in Fig. 6. The energy gap between MOs is 7.90 ev in A and B conformers, 7.85 ev in C and D conformers, and 7.65 ev in E and F conformers, suggesting that the molecule is slightly soft in the latter form. This is confirmed by their larger electronic distribution in LUMO orbitals compared to the other conformers.

Pictorial separation of electronic energy levels with frontier MOs and molecular electrostatic potential plots of 2,2,4-trimethylpentane conformers
Knowing that the red color shows the lowest MEP value (negative) and the blue color the highest one (positive), while the yellow color indicates intermediary potential [41], we noticed that the electrostatic molecular potential also presented in Fig. 6 is almost identical for all conformers, as the yellow color concerns all the skeleton carbon atoms and blue color concerns all hydrogen atoms.
Natural bond orbital analysis
The E(2) stabilization energy indicates the direct relationship between the intensity of ICT and the interaction between bonding and antibonding orbitals. The calculated values of E(2) presented in Table S1 (Supplementary information) have several particularities:
All electronic transfers took place between σ type molecular orbitals.
The E(2) high values reflecting high ICT between bonding orbital σab and antibonding orbital σ*cd concern the ab and cd bonds in trans position.
The larger values of E(2) concern the σCH–σ*CC interactions, especially for σ(C3–H12)–σ*(C2–C8) (B and E conformers) and σ(C3–H13)–σ*(C1–C2) (A and F conformers) as they exceed 4 kcal mol–1.
σ(C3–H12)–σ*(C4–C6) and σ(C3–H13)–σ*(C4–C5) lead large values of E(2) both for E and for F conformers as C4–C6 and C4–C5 are in trans position vis-a-vis of C3–H12 and C3–H13, respectively.
The interactions σCH–σ*CH and σCC–σ*CH are intense but less intense than the σCH–σ*CC interactions and are localized in the interval of 2 to 4.5 kcal mol–1.
Considering the branching of the iso-octane, we were particularly interested in the intramolecular interaction of carbon skeleton σCC–σ*CC with the aim of differentiating between conformers. We noticed the interaction of σC2C7 of the tert-butyl group with σ*C3C4 for all six conformers A–F had values within the interval of 2.5 to 3 kcal mol–1. It is the same for the interaction of σC4C5 of the isopropyl group with σ*C2C3 having the values 1.42 and 2.76 kcal mol–1 for the conformers A and D, respectively, and σC4C6–σ*C2C3 interaction having the values 1.50 and 2.76 kcal mol–1 for B and C conformers, respectively. It is worth noting that the reverse interaction between the bonding and antibonding orbitals is correspondingly diminished.
From E(2) values analysis, it could be concluded that the conformational flexibility affects intramolecular charge transfers of natural bond orbitals.
First-order hyperpolarizability and nonlinear optics analysis
As presented in Table 4, the highest value of dipole moment is observed for A and B conformers, this value is equal to 0.58 Debye. The values of static polarizability or the mean polarizability α0 are very close for the six conformers A–F, while the total polarizability or the anisotropy of the polarizability Δα is relatively lower for the two most stable conformers A–B compared to the secondary ones. The magnitude that is more sensitive to conformers is the first-order hyperpolarizability (β0). Indeed, the B3LYP/6-311++G(d,p) first-order hyperpolarizability values are, respectively, 466.452 10−33 esu and 476.250 10−33 esu for A and B conformers, 601.951 10−33 esu and 603.320 10−33 esu for C and D conformers, and 789.880 10−33 esu and 793.270 10−33 esu for E and F conformers. β0 of A and B conformers is greater than that of urea, taken as a reference [27, 42, 43] of about 27%, while β0 of the secondary conformers C–F is almost two times larger than that of urea leading to moderate nonlinear optics activity of the title molecule.
Carbon 13C and proton 1H NMR spectra prediction
The theoretical relative 13C and 1H-NMR chemical shift values are reported in ppm as shown in Tables 5 and 6 for all the conformers and illustrated for the most stable conformer A (Fig. 7). They were also correlated with natural NPA charges and compared with the experimental data where the skeleton carbons are overlapping with chemical shift values from 25 to 55 ppm and hydrogens from 0.89 to 1.65 ppm [44, 45].

The experimental and calculated 13C and 1H-NMR chemical shifts (ppm) of the most stable conformer A
We first observed the high value exceeding 50 ppm of the central carbon C3, the point of connection between the tert-butyl and isopropyl groups. In the same direction, with a value of about 40 ppm, δC2 of central tertiary butyl carbon exceeds those of the connected peripheral carbons (C1, C7, and C8), and with a value ranging from 30 to 35 ppm, δC4 of central isopropyl carbon exceeds those of the connected peripheral carbons (C5 and C6). These results show clearly that the electronic depletion concerns the central skeletal carbons. Indeed, the natural NPA atomic charge of skeletal carbons varies from −0.1 to −0.4, whereas it is of the order of −0.6 for all peripheral carbons (Tables 5 and 6). We also noted the slightly elevated value of δC7 compared to the other peripheral carbons due to its coplanarity with the C2, C3, and C4 carbons, giving its electrons more mobility. On the other hand, the highest 1H-NMR chemical shift (varying from 1.5 to 1.9 ppm going from A to F) was found for H14, which is bound to the isopropyl C4 carbon, is correlated with the lowest natural charge (0.184) that is less than the other charges by about 0.014.
Knowing that the different conformers are differentiated by the configuration of the isopropyl group, we noted that the chemical shifts δC5 and δC6 are of the order of 26 ppm for the conformers A and B while they exchange their values from C (δC5 = 22 and δC6 = 26 ppm) to D and from E (δC5 = 22 and δC6 = 24 ppm) to F, with a higher chemical shift affecting the carbon that is closer to the plane C7C2C3C4. In addition, the H14 of the isopropyl group is also a source of differentiation, since the value of its chemical shift is respectively of the order of 1.5, 1.7, and 1.9 ppm for (A, B), (C, D), and (E, F).
Moreover, the experimental values of δC5, δC6, and δH14 are more compatible with the conformers A and B than the C, D, E, and F conformers, whereas the calculation of the correlation between observed and theoretical chemical shifts, particularly in 13C-NMR (Fig. 8, Table 7), rejected categorically the conformers E and F as stable conformers. Indeed, they are not only of lower stability but also require a relatively high barrier of rotation.

The observed and calculated 13C-NMR chemical shifts correlation of all A–F conformers
Electronic UV spectra
Time-dependent density functional theory (TD-DFT) is calculated on the optimized geometries by using the same basis sets and hybrid functional, in order to extract the ground to excited state transitions, the excitation energies, and oscillator strengths, with respect of the Franck–Condon principle [46].
The TD-DFT electronic transitions of iso-octane was calculated without experimental results. Indeed, the saturated form of this molecule gives it a high HOMO–LUMO energy gap (almost 8 ev), making it more stable and the σ–σ* electronic transition more difficult (shorter wavelength of absorption maxima).
The excitation wavelengths (λ), energies (E), oscillator strengths (f), and the assignment of electronic excitation of each isolated iso-octane conformer (A–F) and in solvent DMSO are presented in Table 8, while the theoretical electronic spectrum is shown in Fig. 9.

Theoretical electronic spectrum (oscillator strengths (au) as a function of absorption wavelength (λ) (nm)) in gas phase (a) and in DMSO solvent (b) for all A–F conformers
The results of conformers A and B disclose three relatively intense electronic transition peaks at 163, 167, and 171 nm, with the respective values of the oscillator strength 0.081, 0.044, and 0.062 assigned respectively to HOMO-2 -> LUMO, HOMO-1 -> LUMO and HOMO -> LUMO excitation, as main contributions. The conformers C and D exhibit the same main contributions with a slight shift not exceeding the unit for (λ) and 0.2 for (f), compared to A and B conformers. However, the transition HOMO-1 -> LUMO has been submitted to a (λ) shift of 2 nm, from C or D to E or F, while specifically for these latters the transition HOMO -> LUMO+1 appears (contribution of about 70%) at 165 nm with (f) about 0.05 au.
On the other hand, the use of DMSO solvent has a noticeable effect on (f) which increases by about 20 to 25% for all the conformers in addition to the reappearance of the HOMO -> LUMO excitation at 177 nm even with weak strength of about 0.02, and the disappearance of HOMO-2 ---> LUMO transition, for E and F conformers.
Temperature sensitivity and scaled vibrational assignment of conformers
As it is commonly accepted that the conformational behavior of Raman and IR spectra is very sensitive to temperature, mainly at low frequencies, we recorded, in our earlier work [47], the Raman spectrum of iso-octane in liquid phase below 600 cm−1 in the temperature range from 293 to 183 K. As is shown in Fig. 10, if we exclude the refinement of bands caused by cooling of the sample, we did not notice any exchange between bands in terms of intensity, thereby removing any possibility of conformational change. It should be noted that the infrared spectrum between 200 and 600 cm −1 does not show any bands that are inactive in the Raman spectrum. For this reason, temperature sensitivity of the infrared region was not considered.

Observed low frequency Raman spectrum of 2,2,4-trimethylpentane, in liquid phase below room temperature
The absence of temperature-sensitive bands in the spectra of this isomer can be related to the fact that the vibrational frequencies of the two most stable conformers A and B are not sufficiently shifted from the secondary ones. To confirm this, the scaled vibrational normal modes calculations were made leading us to the harmonic frequencies of vibrations adjusted to the observable frequencies as well as the distribution of the potential energy for each calculated normal mode. To highlight the local symmetry of the carbon skeleton, we considered the symmetry coordinates based on the C3v local symmetry of the tert-butyl group (DS TS DD SD DR CCC), while the C2v symmetry of the isopropyle group is ignored because the rotation axis does not coincide with the CC rotation. All used symmetry coordinates are derived from internal coordinates and detailed in Table S2 (Supplementary information).
The refinement of the ab initio normal modes frequencies has been carried out through the optimization of the scale factors of B3LYP/6-311++G(d,p) force constants for the conformer A (Table 9). For the other less stable conformers, the variation of scale factors compared to A does not exceed 4%. Raman and IR frequencies, in liquid phase, as well as the adjusted calculated frequencies are listed in Table 10. As shown by the temperature sensitivity study of the Raman low frequency region, eliminating any possibility of conformational exchange, all observed frequencies are predicted by scaled normal mode calculation for the most stable conformer A. Thus, only the PED for conformer A is given, all minor contributions (contributions <10%) are eliminated unless they consolidate the preponderant contribution, in this case, even the contributions going down to 5% are considered. On the whole, the computed scaled frequencies are in good agreement with the experimental data leading rms deviation, not exceeding 10 cm−1 for all frequencies and 6 cm−1 below 1500 cm−1. The comparison between calculated and observed intensities was used, mainly in Raman, to verify the symmetrical character of the calculated mode of vibration. Indeed, the more symmetrical the mode, the higher its Raman intensity.
As the molecule 2,2,4-trimethyl pentane contains five CH3, one CH2, and one CH, its different vibration modes are subdivided into two groups. The first group (18 modes) contains ten degenerates asymmetric stretching CH3ds (in plane and out of plane), five symmetric stretching CH3ts, one asymmetric stretching CH2as, one symmetric stretching CH2ss, and one CH stretching CHs. All these modes are considered pure as long as the complementary contributions are minor and do not exceed 10%. Their respective frequencies are observed and well predicted in the region [2900, 2840] in accordance with the following order: CH3ds > CH2as > CH3ts > CH2ss > CHs, as found in our earlier work and those of Mirkin et al. for some normal and congested alkanes [48]. The second group contains, as pure modes or in combination, ten degenerate asymmetric deformations CH3ab (in plane and out of plane), five symmetric deformations CH3sb, five degenerate rocking CH3dr (in plane and out of plane), one CH2sc scissoring, one CH2wa wagging, one CH2tw twisting, one CH2ro rocking, CCH defCH, and CCC deformations. As can be seen from the B3LYP/6-311++G(d,p) scaled ab initio vibrational computations for the conformer A, CH3ab was located as pure mode or combined with CH2b from 1480 to 1445 cm−1 and observed from 1470 to 1450 cm−1, while CH3sb appears essentially alone from 1390 to 1360 cm−1 and calculated in the same order of magnitude. HCC deformation DefCH of the tertiary carbon C4 appears alone for two modes calculated at 1351 and 1348 cm−1 and observed at 1350 cm−1 and contributes with CH2wa, CH2tw or CC stretching for two other modes calculated at 1299 to 1280 cm−1 and observed at the same frequencies.
Furthermore, the CC stretching acts within the range 1250–740 cm−1, contributes essentially in combination with CH2ro, CH2tw, and CH3r, knowing that the most important modes involving predominately CC contributions were calculated at 1248 (40%), 896 (48%), 832 (55%), and 747 (74%) and observed at 1245, 897, 831, and 748 cm−1, respectively. The latter band is very strong and polarized [49] in Raman spectrum as is assigned to total symmetric stretching (TS) of the tert-butyl group (60%) and C2C3 stretching (14%). Concerning the CCC deformation, it appears predominately in the 510–350 cm−1 interval or combined with CC torsions below 350 cm−1 knowing that the deformation mode of the central skeleton contributes predominately to C2C3C4 (50%) and the two torsional modes involving predominately C2C3 and C3C4 torsions have the lowest frequencies of CCC deformation and CC torsion, respectively.
In order to complete our conformational analysis, the normal modes of secondary conformers based on the same B3LYP/6-311++G(d,p) scaled ab initio force field as the conformer A were determined. We noticed that all the observed frequencies were reproduced by the vibrational mode calculation of B, C, D, E, and F conformers, indicating once more, that all observed bands are common for the most stable conformers as well as the secondary ones.
Conclusions
In the light of this large theoretical and comparative study, we can conclude the following:
A large exploration of the conformational space has led us to two very stable conformers and four other secondary ones, where the interaction between tert-butyl and isopropyl groups was a determining factor.
All possible transitions between conformers are one-dimensional rotations around the C3–C4 bond and are confronted with isopropyl barriers to internal rotation.
The B3LYP/6-311++G(d,p) optimization is slightly more stabilizing than MP2/6-311++G(d,p) and MP2/6-311++G(d,p) rotational barriers, and their corresponding inversion barriers between secondary conformers are larger than that of B3LYP/6-311++G(d,p).
The secondary conformers E and F are slightly softer, and thus more reactive than the other forms, while the electrostatic molecular potential is almost identical for all conformers.
According to E(2) values, the conformational flexibility influences intramolecular charge transfers of natural bond orbitals and the larger values of E(2) are explained with the σCH–σ*CC interactions.
The magnitude of the first-order hyperpolarizability is found to be very sensitive to conformational behavior, contrary to mean polarizability and the anisotropy of the polarizability. However, the title molecule has only moderate nonlinear optics activity.
The theoretical relative 13C and 1H-NMR chemical shift predictions confirmed their conformational sensitivity, by their compatibility with natural NPA charges. In addition, their confrontation in the experiment confirms that the conformers E and F are not only of lower stability but also require a relatively high barrier of rotation.
Only the least stable secondary conformers E and F are easily distinguishable from the other conformers by TD-DFT calculations on electronic absorption spectra and the use of DMSO solvent has a noticeable effect on oscillator strengths, which increase by about 20 to 25% for all the conformers.
Spectral temperature sensitivity and scaled vibrational assignment for the six lowest conformers A–F are in a good agreement, as both procedures eliminate any possibility of conformational exchange between conformers.
The computed scaled frequencies are well predicted as they are in a good agreement with the experimental data rms deviation, not exceeding 10 cm−1 for all frequencies and 6 cm−1 for frequencies below 1500 cm−1.
As a perspective of this work, we plan to extend our study to comparable molecules, especially the other molecules in the trimethylpentane series, with more advanced quantum methods.
References
Dabelstein W, Reglitzky A, Schutze A, Reders K (2012) Automotive fuels. In: Ullmann’s encyclopedia of industrial chemistry. Wiely-VCH, Weinheim, pp 426–457
Frédéric B (2006) Mécanismes cinétiques pour l’amélioration de la sécurité des procédés d’oxydation des hydrocarbures. Dissertation, University of Nancy
Chiari L et al (2014) Cross sections for positron impact with 2,2,4-trimethylpentane. J Phys Chem A 118:6466–6472
National Center for Biotechnology Information (2018) 2,2,4-Trimethyl pentane – compound summary. PubChem Compound Database. CID=10907, https://pubchem.ncbi.nlm.nih.gov/compound/10907. Accessed 2 March 2018
Enech OC (2011) A review on petroleum: source, uses, processing, products and the environement. J Appl Sci 11(12):2084–2091
Suvitha A, Periandy S, Govindarajan M, Gayathri P (2014) Vibrational analysis using FT-IR, FT-Raman spectra and HF - DFT methods and NBO, NLO, NMR, HOMO-LUMO, UV and electronic transitions studies on 2,2,4 trimethyl pentane. Spectrochim Acta A Mol Biomol Spectrosc 138:900–912
U.S. Environmental Protection Agency (1999) Integrated Risk Information System (IRIS) on 2,2,4-trimethyl pentane. National Center for Environmental Assessment, Office of Research and Development, Washington, DC
Guntram R, Peter P (1995) Transferable scaling factors for density functional derived vibrational force fields. J Phys Chem 99:3093–3100
Cox SR, Williams DE (1981) Representation of the molecular electrostatic potential by a net atomic charge model. J Comput Chem 2(3):304–323
Reed AE, Weinhold F (1983) Natural bond orbital analysis of near-Hartree–Fock water dimer. J Chem Phys 78(6):4066–4073
Keeler J (2011) Understanding NMR spectroscopy. Wiley, New York
Owen AE (1996) Fundamentals of UV-visible spectroscopy. Hewlett-Packard Company, Palo Alto
Mouatarif S, Aboulmouhajir A (2004) An ab initio and DFT study of the conformational stability in branched alkanes: illustration for 3 , 3-dimethylhexane. J Mol Struct 709:157–161
Werner HJ, Knowles PJ, Knizia G, Manby FR, Schütz M (2012) Molpro: a general-purpose quantum chemistry program package. WIREs Comput Mol Sci 2:242–253
The Magrid Virtual Organization of the Moroccan Grid Infrastructure (2016) CNRST/MAGRID. http://www.magrid.ma. Accessed 20 Apr 2016
Ayala PY, Scuseria GE (1999) Linear scaling second-order Moller–Plesset theory in the atomic orbital basis for large molecular systems. J Chem Phys 110:3660–3671
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Krishnan R et al (1980) Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J Chem Phys 72:650–654
Mouatarif S, Van Alsenoy C, Aboulmouhajir A (2008) Conformational dependence of vibrational spectra in some branched octanes: 3,3- and 2,2-dimethylhexanes. Spectrosc Lett 41:87–99
Zhurko G, Zhurko D (2017) ChemCraft, version 1.8. http://www.chemcraftprog.com. Accessed 04 May 2018
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2010) Gaussian 09, revision B.01. Gaussian, Inc., Wallingford
Alecu IM, Zheng J, Zhao Y, Truhlar DG (2010) Computational thermochemistry: scale factor databases and scale factors for vibrational frequencies obtained from electronic model chemistries. J Chem Theory Comput 6:2872–2887
Martin JML, Van Alsenoy C (2007) GAR2PED, a program to obtain a potential energy distribution from a Gaussian archive record. University of Antwerp, Antwerp
Keresztury G et al (1993) Vibrational spectra of monothiocarbamates-II. IR and Raman spectra, vibrational assignment, conformational analysis and ab initio calculations of S-methyl-N,N-dimethylthiocarbamate. Spectrochim Acta A Mol Biomol Spectrosc 49:2019–2026
Keresztury G, Chalmers JM, Griffith PR (2002) Raman spectroscopy: theory. Hand book of vibrational spectroscopy. Wiley, New York
Grabowski ZR, Rotkiewicz K, Rettig W (2003) Structural changes accompanying intramolecular electron transfer: focus on twisted intramolecular charge-transfer states and structures. Chem Rev 103:3899–4032
Vijayachamundeeswari SP, Yagna Narayana B, Jone Pradeepa S, Sundaraganesan N (2015) Vibrational analysis, NBO analysis, NMR, UV-VIS, hyperpolarizability analysis of Trimethadione by density functional theory. J Mol Struct 1099:633–643
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev 88:899–926
Natarajan S, Shanmugam G, Dhas SAMB (2008) Growth and characterization of a new semi organic NLO material: L-tyrosine hydrochloride. Cryst Res Technol 43:561–564
Avci D (2011) Second and third-order nonlinear optical properties and molecular parameters of azo chromophores: semiempirical analysis. Spectrochim Acta A Mol Biomol Spectrosc 82:37–43
Avci D, Basoglu A, Atalay Y (2010) Ab initio HF and DFT calculations on an organic non-linear optical material. Struct Chem 21:213–219
Altürk S, Avcı D, Tamer Ö, Atalay Y (2017) Comparison of different hybrid DFT methods on structural, spectroscopic, electronic and NLO parameters for a potential NLO material. Comput Theor Chem 1100:34–45
Colherinhas G, Fileti EE, Malaspina T (2018). J Mol Model 24:181. https://doi.org/10.1007/s00894-018-3719-3
Iozzi MF, Mennucci B, Tomasi J, Cammi R (2004) Excitation energy transfer (EET) between molecules in condensed matter: a novel application of the polarizable continuum model (PCM). J Chem Phys 120:7029–7040. https://doi.org/10.1063/1.1669389
Guelai A, Brahim H, Guendouzi A et al (2018) Structure, electronic properties , and NBO and TD-DFT analyses of nickel (II), zinc (II), and palladium (II) complexes based on Schiff-base ligands. J Mol Model 24:301. https://doi.org/10.1007/s00894-018-3839-9
Marques MAL, Gross EKU (2003) Time-dependent density functional theory. In: Fiolhais C, Nogueira F, Marques MAL (eds) A primer in density functional theory. Lecture notes in physics, vol 620. Springer, Berlin
Pauzat F, Talbi D, Miller M, Defrees D, Ellinger Y (1992) Theoretical IR spectra of ionized naphthalene. J Phys Chem 96:7882–7886
Lide Jr DR, Mann DE (1958) Microwave spectra of molecules exhibiting internal rotation. IV. Isobutane, tertiary butyl fluoride, and trimethylphospine. J Chem Phys 29:914. https://doi.org/10.1063/1.1744611
Pitzer KS, Kilpatrick JE (1946) The entropies and related properties of branched paraffin hydrocarbons. Chem Rev 393:435–447. https://doi.org/10.1021/cr60124a005
Jha O, Yadav TK, Yadav RA (2017) Comparative structural and vibrational study of the four lowest energy conformers of serotonin. Spectrochim Acta A Mol Biomol Spectrosc 173:307–317
Shankar Rao YB, Prasad MVS, Udaya Sri N, Veeraiah V (2016) Vibrational (FT-IR, FT-Raman) and UV-visible spectroscopic studies, HOMO-LUMO, NBO, NLO and MEP analysis of benzyl (imino (1H-pyrazol-1-yl) methyl) carbamate using DFT calculaions. J Mol Struct 1108:567–582
Sasikala V, Sajan D, Chaitanya K, Sundius T, Devi TU (2017) Qualitative and quantitative approach towards the molecular understanding of structural, vibrational and optical features of urea ninhydrin monohydrate. Mater Chem Phys 191:20–34
Sun YX et al (2009) Experimental and density functional studies on 4-(3,4-dihydroxybenzylideneamino)antipyrine, and 4-(2,3,4-trihydroxybenzylideneamino)antipyrine. J Mol Struct THEOCHEM 904:74–82
National Institute of Advanced Industrial Science and Technology (AIST) (1999) https://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_disp.cgi?sdbsno=2353&spectrum_type=CNMR&fname=CDS00677. Accessed 16 April 2019
National Institute of Advanced Industrial Science and Technology (AIST) (1999) https://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_disp.cgi?sdbsno=2353&spectrum_type=HNMR&fname=HSP03717. Accessed 16 April 2019
Condon E (1947) The Franck-Condon principle and related topics. Am J Phys 15:365–374. https://doi.org/10.1119/1.1990977
Aboulmouhajir A, Mouatarif S et al (2017) Theoretical and spectroscopic investigations of conformations, rotational barriers and scaled vibrations of 2 , 3-dimethyl hexane. Mediterr J Chem 6:60–70
Mirkin NG, Krimm S (2000) Ab initio analysis of the vibrational spectra of conformers of some branched alkanes. J Mol Struct 550–551:67–91
Gough KM, Lupinetti C, Dawes R (2004) Computation and interpretation of Raman scattering intensities. J Comput Methods Sci Eng 4:597–609
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 196 kb)
Rights and permissions
About this article

Cite this article
Hachim, M.E., Sadik, K., Byadi, S. et al. Ab initio study on the six lowest energy conformers of iso-octane: conformational stability, barriers to internal rotation, natural bond orbital and first-order hyperpolarizability analyses, UV and NMR predictions, spectral temperature sensitivity, and scaled vibrational assignment. J Mol Model 25, 254 (2019). https://doi.org/10.1007/s00894-019-4105-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-4105-5