
Abstract
Our study examines the mechanisms by which DNA polymerase (pol) δ faithfully replicates DNA. To better understand this process, we have performed all-atom molecular dynamics simulations of several DNA pol δ systems to identify conformational changes occurring prior to chemistry and investigate mechanisms by which mutations in the fingers domain (R696W and A699Q) lower fidelity. Our results indicate that, without the incoming nucleotide, a distinct open conformation occurs defined by a rotation in the fingers. The closed form, adopted when the correct nucleotide is bound, appears best organized for chemistry when three magnesium ions coordinate protein and DNA residues in the active site. Removing an unusual third metal ion from the polymerase active site causes shifting in the fingers and thumb as well as stimulating specific exonuclease β-hairpin—DNA interactions that fray the primer terminus base pair. These changes suggest that dissociation of the third divalent ion (metal ion ‘C’) signals a transfer of the DNA primer from the polymerase to the exonuclease active site and implies a role for the β-hairpin in DNA switching. Analysis of β-hairpin movement in several systems reveals a dependence on active-site changes and suggests how Lys444 and Tyr446 present in the β-hairpin can affect proofreading. Analysis of A699Q and R696W pol δ mutant systems reveal marked differences in the open-to-closed transition as well as β-hairpin repositioning that explain reduced nucleotide selectivity and higher error rates.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The replication of an organism is dependent on the faithful copying of the genomic material in eukaryotic cells. This genomic information is encoded in DNA. Even for a simple organism, such as Saccharomyces cerevisiae with over 12 million base pairs to be copied, this is a daunting task [1]. In humans, by contrast, there are approximately 3 billion base pairs [2, 3]. DNA replication is critically dependent on three eukaryotic DNA polymerases: α, δ, and ε [4]. During replication, a DNA polymerase (pol) extends a growing primer strand by catalyzing a nucleotidyl transfer reaction in which the O3′ atom of the primer terminus attacks the Pα atom of an incoming nucleotide (dNTP, 2′-deoxyribonucleoside 5′-triphosphate). DNA pol α is a multi-subunit enzyme that synthesizes short RNA-DNA primers through primase and DNA polymerase activities, while pols δ and ε handle the majority of the copying task by further elongating the primers [5]. Experimental evidence suggests that pol ε primarily replicates the leading strand and pol δ the lagging strand, but the interplay of these three polymerases in completing the task of DNA replication remains an area of active research [6,7,8,9,10,11,12,13]. In addition to DNA replication, pol δ also functions within several DNA repair pathways including mismatch repair, base excision repair, nucleotide excision repair and homologous recombination double-strand break repair [14]. Recent research suggests that pol δ may also play a role in telomere lengthening under certain circumstances [15].
The fidelity of DNA replication is achieved largely by the effectiveness of pols δ and ε in selecting the correct incoming nucleotide over an incorrect nucleotide to faithfully extend a growing primer strand [16, 17]. Correct nucleotides form Watson-Crick base pairs (guanine-cytosine and adenine-thymine) that allow the DNA helix to maintain a regular helical structure. The base substitution error rate of yeast pol δ is less than 1.3 base substitutions per 100,000 nucleotides incorporated [18], and the error rate of human pol δ is approximately 3-fold lower [19]. To help reduce replication errors, pols δ and ε possess 3′-5′ exonuclease activity that allows these enzymes to excise incorrect nucleotides that are inserted. The proofreading ability of pol δ also can correct errors made by pols α and ε [20, 21].
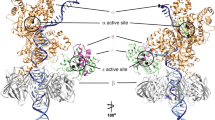
Pols α, δ, and ε belong to the B-family of polymerases, which also includes bacteriophage T4 and RB69 polymerases as well as translesion polymerases such as eukaryotic pol ζ (or REV3) and Escherichia coli Pol II [22, 23]. Several X-ray crystal structures are available for this family of enzymes [24,25,26,27,28,29,30,31,32,33], including one structure of pol δ shown in Fig. 1. B-family polymerases adopt the shape of a right hand, and consist of five domains: fingers, palm, thumb, exonuclease domain, and an N-terminal domain (NTD) [31]. The thumb grasps the DNA, the fingers help position the incoming nucleotide in the active site, and the palm is the site of the nucleotidyl transfer reaction. The exonuclease domain provides the enzyme with proofreading ability, allowing it to excise incorrect nucleotides. The NTD, positioned between the exonuclease and fingers domains, and in contact with the single-strand portion of the DNA template strand, may play a role in stabilizing the overall polymerase-DNA complex.

DNA polymerase δ/DNA/dCTP complex (dCTP, 2′-deoxycytidine 5′-triphosphate). a Protein colored by protein domain. Colors of DNA and ions are indicated on image. b Solvated complex used for simulations. Water is shown as blue background. c Positions of Ala699 and Arg696 in the fingers domain
Rearrangements in these protein domains are part of the replication process. Conformational changes are often interpreted in terms of an induced-fit mechanism, where binding of the correct nucleotide facilitates the transition to the closed form needed for the chemical reaction, while binding of an incorrect nucleotide hampers this transition [34]. Open-to-closed conformational changes have been identified in the catalytic cycles of several polymerase systems, including pol β [35, 36], T7 pol [37, 38], pol I [39, 40], and HIV RT [41, 42], and computer simulations have helped reveal the dynamic details of these conformational rearrangements [43,44,45,46,47,48,49,50,51]. For the B-family, a structural comparison of RB69 pol in the apo or unliganded state to the ternary pol/DNA/dNTP state shows that rotations in the fingers and thumb produce a closed conformation when the DNA and correct incoming nucleotide are bound [25, 52]. Similar changes occur in E. coli Pol II [27] and yeast pol α [28].
Interestingly, there is a diversity in the conformational changes occurring in polymerase catalytic cycles, especially among DNA repair polymerases [53,54,55,56]. In the B-family, structural data indicate that the magnitude of the conformational closing change varies. For instance, pol α has a larger thumb rotation compared to RB69 pol [28]. Additional conformational changes are associated with the polymerase proofreading process. In RB69 pol, a distinct thumb shift occurs to facilitate the positioning of the DNA primer strand in the exonuclease active site to form the exonuclease complex; thus, the thumb position changes between polymerizing and editing functional modes [25, 57].
Elucidating the conformational changes associated with the proper assembly of the active site is a primary goal of this work. As shown in Fig. 1, the available pol δ structure is of a ternary pol δ/DNA/dNTP complex [26]. Pol δ tightly surrounds the DNA by making extensive contacts with several DNA base pairs, which may help the polymerase detect irregularities in the DNA conformation resulting from binding mismatches or misaligned DNA. The fingers and thumb resemble the closed conformations of other B-family enzymes. In the active-site pocket, the correct nucleotide is bound along with three divalent ions coordinated by three carboxylic acid containing residues. The presence of a third ion in the active site is unusual since most DNA polymerases are hypothesized to use a two-metal-ion nucleotidyl transfer mechanism [22]. In this mechanism, the catalytic ion or metal ion ‘A’ lowers the pKa of the primer terminus’ 3′-OH to prepare the O3′ atom for attack on the Pα atom of the dNTP, while the nucleotide-binding ion or metal ion ‘B’ facilitates formation of the pyrophosphate product. Both of these ions are proposed to stabilize the charge and geometry of the transition state. In pol δ, the importance of the third divalent ion or metal ion ‘C’ is unknown, but biochemical studies suggest it has a role in the catalytic efficiency of the enzyme [26]. Recently, X-ray crystal structures of an archaeal B-family polymerase also show metal ion ‘C’ bound to the active site in closed ternary complexes, reinforcing the importance of this third metal ion [58]. Although binding in a different manner, a third metal ion is observed in the active sites of both Y-family pol η [59, 60] and X-family pol β [61, 62], and is proposed to be necessary for catalytic activity [60, 62].
Gaining mechanistic details of proofreading activity is needed to have a full view of accurate DNA replication. Pol δ’s proofreading exonuclease domain has a β-hairpin positioned near the DNA major groove that is conserved in many B-family polymerases. This protruding β-hairpin in the RB69 and T4 pols is proposed to facilitate the separation of the primer and the template strands, and thus assist switching of the DNA between polymerase and exonuclease active sites, which are located ~40 Å apart [57, 63,64,65,66,67]. Failure to efficiently transfer DNA to the exonuclease active site favors mismatch extension and results in error-prone replication. Despite the structural similarity of the β-hairpin in B-family polymerases, hairpin length and amino acid sequences vary. In addition, the roles of even conserved residues appear to differ in B-family polymerases as indicated by the differing effects on fidelity of mutating a conserved glycine residue in T4 pol, RB69 pol, and pol δ [63, 66, 68, 69]. Furthermore, from studies of chimera enzymes, it is evident that the β-hairpins do not function identically in different polymerase systems: pol δ with the β-hairpin of T4 pol displays increased mutations while the pol δ β-hairpin on T4 pol is incompatible with cellular survival [69]. Since pol ε has a much shorter β-hairpin [29, 30], it could not employ the same mechanism for switching DNA to the exonuclease domain [70]. These findings have led to the proposal that each polymerase β-hairpin has evolved a specific function, with the degree of participation in proofreading dependent upon whether the organism possesses a mismatch repair pathway to help with this process [69].
Due to pol δ’s multiple cellular roles, it is likely that functional changes that increase errors would be highly deleterious. Indeed, mutations in pol δ’s exonuclease domain are predisposition markers for a class of hereditary colorectal and endometrial cancers in humans [71, 72]. Also, a mutation in pol δ’s polymerase domain, R689W, occurs in a human colon cancer cell line [73]. This particular mutant maps to the fingers domain, and the analogous mutation in yeast, R696W, has reduced polymerase activity and nucleotide selectivity [74, 75]. As a result, the R696W mutant has an ~750-fold higher mutation rate [75]. Another change in the fingers (i.e., A692Q in humans and A699Q in yeast) has been found to lower fidelity [76]. This alanine to glutamine mutation leads to an ~20-fold higher mutation rate in yeast and an ~30-fold higher mutation rate in humans [76]. Base substitution errors commonly result, suggesting a decrease in nucleotide selectivity, but data also suggest the mutant enzyme has an impaired proofreading ability [76]. Thus, these different mutations demonstrate the importance of any changes in the fingers domain for pol δ function and fidelity.
To investigate the unique conformational change mechanism of pol δ and interpret the impact on function and fidelity, we performed atomistic molecular dynamics simulations of several wild-type and mutant pol δ systems (Table 1). Computer simulations provide structural, energetic, and dynamics information for enzyme systems within different substrate contexts [77, 78]. Specifically, we sought to identify conformational changes occurring during the pol δ catalytic cycle and elucidate the factors that promote nucleotide selectivity and proofreading ability. Additionally, we aimed to determine the impact of metal ion ‘C’ on the catalytic cycle and to elucidate the means by which mutations in the fingers (R696W and A699Q) affect function and fidelity.
Our results reveal an open conformation in the binary pol δ/DNA complex and support the formation of a closed conformation in the ternary pol δ/DNA/dNTP complex. The open conformation is produced from rearrangements in the fingers, thumb, and exonuclease domain. A key movement involves the fingers rotating away from the exonuclease domain and toward the palm to help stabilize the primer terminus in the active site. Interestingly, the closed form is only maintained in the ternary complex with three magnesium ions bound to the active site. Pol δ ternary complexes with three calcium ions or two magnesium ions show similarities in the fingers and palm to the open state, suggesting that these complexes are less ready for nucleotide insertion. Our pol δ simulations also have implications regarding the initiation of proofreading activity that connects metal ion ‘C’ dissociation and thumb/DNA shifting as well as movement in a conserved β-hairpin in the exonuclease domain. Our simulations of the R696W and A699Q pol δ mutant systems reveal conformational changes that provide explanations for the distinctive functional and fidelity profiles of these enzymes.
Experimental methods
Initial models
All six in silico models summarized in Table 1 are derived from X-ray crystal data for a ternary yeast pol δ/DNA/dCTP complex (PDB ID: 3IAY; dCTP, 2′-deoxycytidine 5′-triphosphate) [26]. This pol δ structure includes the polymerase domain, exonuclease proofreading domain, and an N-terminal domain. In all but one of the models, each divalent calcium ion is replaced with a magnesium ion. This is done since calcium inhibits pol δ activity [26] and magnesium is hypothesized to be the biologically relevant ion [22]. One pol δ model with calcium ions is kept for comparison with the magnesium ion systems. To determine the functional importance of the third divalent ion or metal ion ‘C’ in the polymerase active site, this ion is removed from one of the pol δ models. In another pol δ model, all divalent ions and the incoming nucleotide are removed from the polymerase active site to model the binary pol/DNA complex and probe the dependence of protein and DNA conformations on substrate binding. In two of the remaining models, mutations are made to residues in the fingers domain to elucidate the effects of these residues on the function and fidelity of pol δ. In one model, Ala699 is changed to glutamine and, in the other model, Arg696 is replaced with tryptophan.
All changes to atoms and residues are made using the CHARMM program [79, 80]. This includes adding all hydrogen atoms, N-terminal residues 67–94, and a small connective region in the exonuclease domain corresponding to residues 491–496. In addition, a hydroxyl group is added to the 3′ carbon of the primer terminus sugar moiety to model the system in a chemically active form. Acetate ions present in the structure resulting from the crystallization procedure are removed.
Using the VMD program [81], each system is solvated in a water box and sodium and chloride ions are added to neutralize and provide 150 mM ionic strength. The counterions are placed with a separation of at least 5 Å between the ions and between the ions and protein or DNA atoms. In all systems, there are 88 Na+ ions. The WT-3CA, WT-3MG, and A699Q systems (see system labels in Table 1) each contain 66 Cl− ions. The WT-2MG and WT0 systems each have 64 Cl− ions. The R696W system has 65 Cl− ions. As shown in Fig. 1, each fully solvated system contains approximately 100,349 atoms, including 558 crystallographically resolved water molecules and 27,786 bulk water molecules. The final dimensions of each system are: 114.8 Å × 100.9 Å × 84.0 Å.
Minimization, equilibration, and dynamics protocol
The NAMD program [82] with the all-atom CHARMM protein and nucleic-acid force field [83, 84] was used to perform all energy minimization and molecular dynamics simulations. To prepare each system for production phase dynamics, several cycles of system energy minimization and short dynamics simulations were performed during an equilibration phase. This process began by relaxing the water molecules and sodium and chloride ions around the protein/DNA complex. This was carried out by minimizing the energy of each system using the Powell conjugate gradient algorithm for 150,000 steps while all protein atoms, nucleic atoms, and active-site divalent ions are kept fixed. Then, with the same constraints, each system is equilibrated with a 30 ps simulation at 300 K under conditions of constant pressure and constant temperature. Pressure is maintained at 1 atm using the Langevin piston method [85], with a piston period of 100 fs, a damping time constant of 50 fs, and piston temperature of 300 K. Using these same conditions, each system is further energy minimized 75,000 steps, equilibrated for 60 ps and then energy minimized again for 50,000 steps. The water, sodium ions, and chloride ions are further equilibrated at constant volume and temperature for 100 ps at 300 K while holding all other atoms fixed. In the following portion of the equilibration phase, all constraints are removed. This portion includes two cycles of energy minimization and dynamics on the entire system. In the first cycle, the energy is minimized for 50,000 steps followed by equilibration for 100 ps at 300 K at constant temperature and volume. In the second cycle, 25,000 steps of energy minimization are performed, followed by 150 ps of dynamics at 300 K at constant temperature and volume.
Following system equilibration, production dynamics are performed at constant temperature and volume. The temperature is maintained at 300 K using weakly coupled Langevin dynamics of non-hydrogen atoms with damping coefficient, γ, 5 ps−1 used for all simulations performed. A time step of 2 fs is used with the SHAKE algorithm [86] to keep all bonds to hydrogen atoms rigid. The system is simulated in periodic boundary conditions and full electrostatics are computed using the PME method [87] with grid spacing on the order of 1 Å or less. Short-range nonbonded interactions are evaluated every step using a 12 Å cutoff for van der Waals interactions and a smooth switching function. The total simulation length for each of the six systems is 100 ns, which amounts to 0.6 μs of total pol δ simulation time.
Results
Evidence for separate open and closed pol δ conformations
Fingers domain motion produces an inactive chemical state
To identify rearrangements in the fingers of pol δ that are important for function and fidelity, we examined the fingers motion in all pol δ simulations by analyzing the RMSD of finger Cα atoms shown in Fig. 2. Although no significant fingers movement from the closed orientation occurs in the WT-3MG system or the A699Q mutant system, some small changes occur in the WT-3CA and WT-2MG systems as indicated in Fig. 2a–d; see system descriptions in Table 1. In the WT-3CA system, a reversible fingers motion occurs over the last 50 ns of the simulation; this primarily involves α-helix O of the fingers moving toward the palm. In the WT-2MG system, a shift in α-helix O of the fingers toward the palm occurs that strengthens interactions between the fingers and palm (see description in “Open state enables support of the primer terminus by both the fingers and palm” section).

Time evolution of the RMSD of pol δ Cα atoms in the fingers domain in all system trajectories relative to the X-ray crystal structure conformation of pol δ (PDB ID: 3IAY). Superposition is performed with respect to protein Cα atoms from residues 91 to 985. RMSD plots do not start at zero because each system was equilibrated prior to production dynamics. a WT-3MG, b WT-3CA, c WT-2MG, d A699Q, e WT0, f R696W
As shown in Figs. 2e and 3, removing the dCTP and all active-site ions (WT0 system; Table 1), leads to a shift in the fingers domain away from the DNA to produce an open conformation. In this open conformation, α-helix P of the fingers moves away from the exonuclease domain and adopts a less rigid position, while α-helix O in the fingers moves closer to the palm domain (see Fig. 3a and distance plots in Fig. S1A,E in the Supporting Information). The α-helix O change is similar to that observed in the WT-3CA and WT-2MG systems.

Characterization of the opening motion in pol δ’s fingers domain. a The closed fingers position in the ternary pol δ complex (PDB ID: 3IAY, green) compared with the open positions in the WT0 (red, at 78.4 ns) and the R696W mutant (blue, at 67.1 ns) systems. Distances shown are between the Cα atoms of exonuclease domain residue Phe486 and fingers residue Phe688. The DNA (silver) and calcium ions (orange) are from the pol δ ternary complex. b Magnitude of rotation in pol δ’s α-helix P of the fingers to the open position of analogous helices in RB69 pol (apo form, PDB ID: 1IH7, purple) and human pol α (binary complex, PDB ID: 5IUD, orange)
Interestingly, an opening movement of the fingers also occurs in the R696W system (Table 1), which contains the correct nucleotide and three active-site ions, as summarized in Figs. 2f and 3. In the R696W system, α-helix P rotates away from the exonuclease domain (Fig. S1B in the Supporting Information), but the presence of the dNTP and ions in the active-site pocket prevents a reduction in the distance between the palm and α-helix O as occurs in the WT0 system. Contrastingly, a widening in the distance between the fingers (α-helix P) and palm occurs (see positions in Fig. 3 and distances in Fig. S1C–F in the Supporting Information). Superimposing the open R696W mutant conformation onto the closed form of pol δ highlights the overall widening in the cleft formed between the palm and the fingers (Fig. S2A in the Supporting Information). This more open conformation facilitates the dissociation of the dCTP and ions, which move with the fingers away from the primer terminus (see Fig. S2B in the Supporting Information and a further description of the active-site changes below in “Comparison of active-site geometries in pol δ systems” section). Together, these changes suggest that the fingers rotation to an open conformation produces an inactive state that is not ready for chemistry.
A comparison of the open fingers position in pol δ to that in the apo form of RB69 pol [25] and the binary pol α/DNA complex [33] indicates that the open-to-closed transition is smaller in pol δ, which consists of a 14° rotation of α-helix P from the closed orientation as shown in Fig. 3. Alignment of the structures of these polymerases shows that the open fingers of pol α and RB69 pol are 21° and 25°, respectively, rotated from the closed pol δ fingers position (Fig. 3b). This agrees with the overall relative changes in the fingers previously noted in pol α and RB69 pol [25, 33]. Thus, the extent of fingers rotation varies among related B-family enzymes.
Fingers movement breaks salt bridge joining α-helices of fingers
Interestingly, in the pol δ systems exhibiting movement in the fingers, a salt bridge connecting the two helices of the fingers breaks or, in the case of the R696W system, is unable to form due to the mutation in the fingers. As shown in Fig. S3 in the Supporting Information, this interaction is between Asp680 from α-helix O and Arg696 from α-helix P. Dissolution of this interaction permits the independent shifting of the fingers helices described above. Simultaneously, Arg696 forms a salt bridge to N-terminal residue Glu539, which remains intact during simulations (Fig. S4 in the Supporting Information). Since Arg696 is the residue mutated in the R696W system, complete removal of both of these salt bridges facilitates opening of the fingers despite the presence of the correct nucleotide in the active site.
Open state enables support of the primer terminus by both the fingers and palm
Stacking interactions between the primer terminus and dNTP as well as coordination to active-site ions help to anchor the DNA to the active site. To identify the support network between the protein and active-site DNA that forms when the dNTP and ions are removed, we examined interactions among the DNA, fingers, and palm in all systems. Due to the relative proximity of the fingers and palm in the closed conformation adopted by the WT-3CA, WT-3MG, and A699Q systems, hydrogen bonds occasionally form between residues in these domains. More frequent hydrogen bonding between the fingers and palm occurs in the WT0, WT-2MG, and R696W systems with the WT0 and WT-2MG systems showing multiple fingers-palm interactions due to shifts in α-helix O of the fingers toward the palm. These hydrogen-bond interactions resemble a series of “gates” to the active site.
As shown in Fig. S5 in the Supporting Information, in the WT0 system, three salt bridge interactions form that involve catalytic residues: Asp764 and Glu802, as well as fingers residue Lys701, which interacts with the triphosphate group of dCTP in the closed ternary complex. The gate closest to the active site joins the primer terminus to Lys701 from α-helix P of the fingers through a common interaction with Asp764 in the palm (see interaction in Fig. S5 and distance data in Fig. S6A,B in the Supporting Information). This fingers-palm-DNA interaction brings Asn705 in α-helix P of the fingers closer to the DNA templating base (Template 4), which provides additional protein/DNA support in the absence of the dNTP (see Fig. S6C in the Supporting Information). The middle gate involves both Glu800 and Glu802 from the palm with Lys678 in α-helix O of the fingers (see Fig. S5 and distance data in Fig. S7A,B in the Supporting Information). The outermost gate joins Asp828 in the palm with Arg690 in α-helix P of the fingers (Figs. S5 and S7C in the Supporting Information). Together, these gates provide a reinforcing network that serves to secure the DNA within the active site.
The WT-2MG system contains the middle and outermost gates (Fig. S7D-F in the Supporting Information). Differently, the middle gate consists of two separate salt bridges: Glu802 (palm)–Lys678 (α-helix O of fingers) and Glu800 (palm)–Arg682 (α-helix O of fingers), instead of both Glu802 and Glu800 coordinating with Lys678 as occurs in the WT0 system. This altered network reflects loss of only metal ion ‘C’ to the active site.
The R696W system, which exhibits a widening in the distance between the palm and α-helix P of the fingers, retains a middle gate only between Glu800 in the palm and Lys678 from α-helix O (Fig. S7G in the Supporting Information). The presence of the dNTP and ions prevents formation of the innermost gate and any interaction between the fingers and Glu802.
Thumb shift causes movement in the DNA primer strand
In pol δ, the thumb makes several contacts with the DNA primer strand upstream of the active site (Fig. 1). These interactions remain during simulations of the WT-3MG, WT-3CA, and A699Q systems. In both the R696W and WT-2MG systems, a shift occurs in the thumb tip (residues 882:952) while the thumb base (residues 835:881 and residues 953:985) remains comparatively unchanged. In the R696W system, movement in the thumb tip follows the opening movement of the fingers. As shown in Fig. 4a, the thumb tip shifts away from the DNA in both systems. The thumb shift is larger in the R696W system than in the WT-2MG system. Specifically, in both systems, the motion involves a thumb loop between β-strand 30 and α-helix U shifting away from the phosphate backbone of the primer strand (Fig. 4a). In the R696W system, another thumb loop adjacent to the DNA that is closer to the active site also shifts away from the primer strand (Fig. 4b). These thumb changes relax the enzyme’s grip on the DNA, allowing a shift of the primer strand in both systems. Due to the changes in both thumb loops in the R696W system as well as the fingers opening, the DNA primer shifting is larger and involves movement in both the primer strand backbone and primer terminus in the active site (Fig. 4b); these changes disrupt stacking interactions between the primer terminus and dCTP. In the WT-2MG system, primer strand movement leads to the fraying of the primer terminus base pair (see “Sustained β-hairpin/DNA template strand interactions cause base pair fraying” section). In the hinge region between the thumb tip and base, residues are closely packed and portions of the α-helices of the thumb base adjacent to the thumb tip show some shifting (see positions of residues in Fig. S8 in the Supporting Information). Together, these changes show that this downward movement of the thumb facilitates movement in the primer strand.

Movement of thumb with respect to the DNA. a Key shifts in parts of the thumb tip occurring in the R696W system (protein, blue) and the WT-2MG system (protein, green) shown after 86.5 ns and 96.6 ns, respectively, of simulation time. For comparison, the crystal position of thumb is shown in orange and DNA in pink (PDB ID: 3IAY). Distances reflect changes from the crystal position. b R696W system changes in thumb loops (residues 835–843 and residues 893–901 near β-strand 30) that contact the primer strand and accompanying shifts in the primer-strand backbone and primer terminus. DNA from the R696W system is shown in red for comparison with crystal DNA position (pink). c Different side-ways shift of the thumb tip in the WT0 system (silver protein shown after 100 ns of simulation time) compared with the crystal position of the thumb tip
Interestingly, a different thumb tip shifting occurs in the WT0 system, as shown in Fig. 4c. This shift follows the helical axis of the DNA upstream from the active site, and this movement of the thumb tip keeps the thumb in close contact with the DNA primer in contrast to the downward motion away from the DNA primer observed in the R696W and WT-2MG systems.
Conformational flexibility of exonuclease domain β-hairpin permits varied interactions with the DNA
β-Hairpin movement overview
B-family polymerases have a conserved β-hairpin in the exonuclease domain that may have a role in proofreading. In pol δ, this β-hairpin, formed between β-strands 16 and 17 in the exonuclease domain, inserts into the major groove of the DNA near the active site. The β-hairpin lies adjacent to exonuclease domain α-helices F and G, which in turn are adjacent to α-helix P of the fingers (see relative positions in Fig. S9 in the Supporting Information).
All pol δ systems except the WT-3MG and WT-3CA systems show significant motion in this β-hairpin (Figs. 5 and 6). In the WT-3MG and WT-3CA systems, the distance between the Cα atoms of Ala445 in the β-hairpin and Arg470 in α-helix F of the exonuclease domain has an average length of ~9 Å based on the distance data shown in Fig. 6a, b. In the WT0 system, this distance can increase substantially (up to ~17 Å; Figs. 5 and 6c). In the WT-2MG and A699Q mutant systems, the β-hairpin is very mobile and the distance between the β-hairpin and α-helix F in the exonuclease domain sometimes decreases significantly (see Figs. 5 and 6d, e). In the WT-2MG system, the distance decreases following shifting of α-helix O in the fingers towards the palm. In the R696W system, the β-hairpin moves further away from α-helix F after the fingers open (compare time of RMSD increase of fingers in Fig. 2f to β-hairpin distance change in Fig. 6f). In the WT-2MG, A699Q, and R696W systems, continued change to the β-hairpin position ceases when the β-hairpin forms new interactions as described in the section “Tyr446 in β-hairpin mediates multiple protein and DNA interactions”.

Exonuclease β-hairpin mobility in pol δ systems. Representative positions of the β-hairpin are shown from the R696W, WT0, WT-2MG, and A699Q simulations to indicate the range of β-hairpin shifting in each system. For comparison, the X-ray crystal structure position of the β-hairpin is shown (PDB ID: 3IAY). Distances are shown between Cα atoms of Ala445 of the β-hairpin and Arg470 of α-helix F. The dCTP is from the X-ray crystal structure

Changes in the position of the exonuclease β-hairpin during all pol δ system simulations. a–f Time evolution of the distance between protein Cα atoms of exonuclease residues Ala445 (from β-hairpin) and Arg470 (from α-helix F) during all trajectories. a WT-3CA, b WT-3MG, c WT0, d WT-2MG, e A699Q, f R696W
Tyr446 in β-hairpin mediates multiple protein and DNA interactions
In the closed form of pol δ, stacking interactions occur between exonuclease domain residues Tyr446 from the β-hairpin and Arg470 from α-helix F as shown in Fig. S9 in the Supporting Information. These interactions persist during simulations of the WT-3MG and WT-3CA systems, where the β-hairpin maintains one orientation (see distance data in Fig. S10A,B in Supporting Information). During simulations of the other pol δ systems, this stacking interaction breaks when the β-hairpin begins moving (compare similar distance change times in Fig. 6c–f and Fig. S10C–F in the Supporting Information).
In the WT0 system, there is no dNTP in the active site, and the β-hairpin is highly mobile. During this simulation, Tyr446 sometimes stacks with Arg470 (Fig. S10C in the Supporting Information), but can also form hydrogen-bond interactions to the DNA due to changes in the β-hairpin position. This includes one hydrogen bond to the primer terminus shown in Fig. S11A in the Supporting Information.
In the WT-2MG system, shifting of the β-hairpin similarly facilitates a hydrogen bond to form between Tyr446 and the primer terminus (Fig. S11B in the Supporting Information). However, a new interaction forms between Tyr446 and Gln469 of α-helix F that hampers further movement of the β-hairpin (Fig. S11B in the Supporting Information). The interaction between these residues is primarily electrostatic in nature and is more energetically favorable than electrostatic interactions occurring between Tyr446 and Arg470 in the WT-3MG system (Fig. 7a, b).

Time evolution of electrostatic interaction energy between Tyr446 and a Arg470 in the WT-3MG system, b Gln469 in the WT-2MG system, and c Gln466 in the A699Q system. To the right of each energy plot are snapshots showing the relative positions of these residues from the last frame of the WT-3MG and A699Q system simulations and after 94 ns of the WT-2MG system simulation
A similar change in the β-hairpin of the WT-2MG system occurs in the A699Q system. When the β-hairpin begins shifting, Tyr446 is repositioned sufficiently close to the primer terminus to form a hydrogen bond (Fig. S11C in the Supporting Information). Continued movement of the β-hairpin facilitates the formation of a hydrogen bond between Tyr446 and Gln466 in α-helix F (Fig. S11C in Supporting Information). The strong electrostatic interaction occurring between Tyr446 and Gln466 restricts movement of the β-hairpin. The interaction between these residues is greater than between Tyr446 and Arg470 in the WT-3MG system and between Tyr446 and Gln469 in the WT-2MG system (Fig. 7).
In the R696W system, when movement of the β-hairpin occurs after the fingers open, Tyr446-Arg470 stacking interactions weaken and break as shown in Fig. S10F in the Supporting Information. The stacking interaction between Tyr446 and Arg470 is disrupted by Arg449, another β-hairpin residue which inserts in between these residues (see Fig. S12 in the Supporting Information). Tyr446 then stacks with Arg449, which also stacks with Arg470. This new arrangement serves to push Tyr446 away from the DNA. No hydrogen bond interaction occurs between Tyr446 and the primer terminus throughout the simulation (Fig. S11D in the Supporting Information). However, Tyr446 in the shifted position occasionally forms a hydrogen bond to the templating guanine nucleotide (Template 4) opposite the dNTP (Fig. S13 in the Supporting Information).
Sustained β-hairpin/DNA template strand interactions cause base pair fraying
In addition to the occasional interactions between Tyr446 and the primer terminus occurring in some systems, exonuclease domain residues Lys444 and Lys473 frequently form hydrogen-bond interactions to the DNA close to the active site in all pol δ systems. Lys444 resides in the same β-hairpin as Tyr446, while Lys473 is located in the loop between α-helices F and G in the exonuclease domain (see residue positions in Fig. S9 in the Supporting Information). In all systems, Lys473 forms hydrogen bonds to DNA-primer backbone oxygen atoms of nucleotides upstream from the active site. Lys444 interacts predominantly with the DNA template strand in all systems, but hydrogen bonds also occur occasionally to the primer strand in the WT-2MG and R696W systems. In the WT-3MG, WT-3CA, WT0, R696W, and A699Q systems, Lys444 forms mainly hydrogen bonds to DNA template strand backbone oxygen atoms. Differently, in the WT-2MG system, Lys444 makes steady hydrogen-bond interactions with DNA template bases (see distance data in Fig. 8) that are facilitated by the new orientation of the β-hairpin. These interactions include hydrogen bonds between the terminal amino group of Lys444 and Template 5:N7 and O6 atoms, which is the template guanine base opposite the primer terminus (Fig. 8a, b). In addition, Lys444’s side chain is within hydrogen-bond distance of Template 4:N7 and O6 atoms (Fig. 8c, d). This “holding” of the DNA template strand by Lys444 facilitates the partial breakage of the primer terminus base pair in the WT-2MG system (Fig. 8e): two of the three hydrogen bonds connecting Template 5 and the primer terminus break.

Interatomic distances between Lys444 and DNA template strand nucleotides 4 and 5 in the WT-2MG system. a, b Proximity between the Lys444 side chain and Template nucleotide 5 (Template 5). c, d Distance between Lys444 and Template nucleotide 4 (Template 4). e Hydrogen bond interaction between Lys444 and Template 5, and the partial breakage of the base pair between Template 5 and the primer terminus after 100 ns of simulation time
A699Q fingers mutation causes repositioning of β-hairpin position and keeps fingers closed
In the A699Q system, mutated residue Gln699 in the fingers is situated close to residues of both the exonuclease domain and the NTD, and causes the side chains of several residues to reposition as depicted in Fig. S14A in the Supporting Information. Specifically, the longer, polar side chain of glutamine compared to alanine destabilizes hydrogen-bond interactions between Tyr537 in the NTD and Glu471 in α-helix F of the exonuclease domain by forming hydrogen-bond interactions to Glu471 (see changes in distances between these residues in Fig. S15 in the Supporting Information). This brings about shifting in α-helix F that disrupts the stacking interaction between Arg470 and Tyr446, and causes the β-hairpin to shift. Following these changes, Gln699 repositions between Met540 and Asn536 of the NTD and forms a hydrogen bond to the backbone oxygen atom of Asn536 as shown in Fig. 9a, b. The stability imposed by this orientation of Gln699 facilitates the return of the Tyr537, Glu471, and Arg470 to their original closed-state positions and a hydrogen bond reforms between Tyr537 and Glu471 (Figs. S14B and S15 in the Supporting Information). The tight fit between these NTD, exonuclease, and fingers residues shown in Fig. 9c appears to lock α-helix P of the fingers into the closed position.

Interactions between Gln699 in the fingers domain and residues in the exonuclease domain and the N-terminal domain (NTD). a, b Position of Gln699 between Met540 and Asn536 of the NTD and hydrogen-bond interactions between the side chain of Gln699 and backbone oxygen atom of Asn536. c Intersection of Tyr537, Met540, Glu471, and Gln699 between α-helix J of the NTD and α-helix P of the fingers domain
Comparison of active-site geometries in pol δ systems
To determine the effect of the above protein and DNA conformational changes on the nucleotidyl transfer reaction catalyzed by pol δ, changes occurring to the active sites of all systems were examined. Figure 10 shows the relative positions of the catalytic residues, ions, primer terminus and dCTP in each system after 100 ns of simulation, and Supplementary Table S1 in the Supporting Information summarizes the key interatomic active-site distances involving the dCTP, primer terminus, bound metal ions, and metal-ion coordinating residues. These data reveal variation in the coordination of calcium compared to magnesium ions in the polymerase active site. In all systems, the catalytic ion ‘A’ is coordinated by six atoms. In both the WT-3CA and WT-3MG systems, these atoms are provided by the primer terminus’ O3′ atom, the dCTP O1α atom, one terminal oxygen of Asp764, and the oxygen atoms of two water molecules. Differently, in the WT-3CA system, catalytic ion ‘A’ is also coordinated by a terminal oxygen of Asp608 while, in the WT-3MG system, a third water molecule coordinates this ion. Coordination of metal ion ‘A’ in the WT-2MG system is similar to the WT-3MG system except that the primer terminus’ O3′ atom alternates with a fourth water molecule.

Organization of the active site in each system after 100 ns of simulation time. The nucleotidyl transfer distance between the O3′ atom of the primer terminus and the Pα of the incoming nucleotide is highlighted in red. The distance between the O3′ atom of the primer terminus and the catalytic ion is shown in blue. Other dashed lines represent coordination of atoms to the divalent ions
In all wild-type pol δ systems, the nucleotide-binding ion ‘B’ is coordinated by both the O2β and O3γ atoms of dCTP, the backbone oxygen atom of Phe609, and one terminal oxygen of both Asp608 and Asp764. Additionally, in the WT-3CA system, the nucleotide-binding ion is coordinated by the O1α atom of dCTP. Very infrequently, a water molecule also coordinates ion ‘B’ in the WT-3MG system.
The coordination of the third metal ion ‘C’ shows the most variability in the wild-type pol δ systems. In both the WT-3CA and WT-3MG pol δ systems, this ion is coordinated by the Oε2 atom of Glu802 and the oxygen atoms of three water molecules. In the WT-3CA system, metal ion ‘C’ is also coordinated by both the O1γ and O3γ atoms of dCTP, although the Oε1 atom of Glu802 briefly substitutes for the O3γ atom of dCTP. Differently, in the WT-3MG system, this ion is coordinated by a terminal oxygen atom of Asp608 and a fourth water molecule.
The A699Q system exhibits the same coordination of all three ions as occurs in the WT-3MG system, but the R696W system only has identical coordination of metal ion ‘C’ to the WT-3MG system. In the R696W system, the coordination of the nucleotide-binding ion ‘B’ is similar to the WT-3MG system, but this ion is also coordinated by dCTP:O1γ and a second terminal oxygen of Asp764 as the fingers open. Coordination of the catalytic ion ‘A’ in the R696W system also changes depending on the open and closed conformations adopted by the fingers: in the closed form, the catalytic ion is coordinated by the primer terminus’ O3′ atom, and, in the open conformation, coordination is substituted by the O1γ atom of dCTP. These ions remain close to the dCTP as the fingers open, bringing about the separation of the primer terminus and dCTP as depicted in Fig. S2B in the Supporting Information.
As indicated by the data in Supplementary Table S1 in the Supporting Information, the coordination of the metal ions in the active site leads to the WT-3CA system having the shortest catalytic distance between the primer terminus’ O3′ atom and the dCTP Pα atom, which form a bond during the nucleotidyl transfer reaction. Interestingly, the WT-2MG system, which does not have metal ion ‘C’, has a longer nucleotidyl transfer distance than the WT-3MG system and increases to ~4 Å. Both the A699Q system and the closed form of the R696W system have nucleotidyl transfer distances similar to the WT-3MG system; however, the open conformation of the R696W system has a catalytic distance 1.7 times longer than the closed form since the dCTP moves away from the primer terminus as the fingers open.
Changes in the distances between metal ions occur for several nanoseconds in the WT-3CA and R696W systems. In the WT-3CA system, as movement in the fingers occurs, the distance between the nucleotide-binding ion ‘B’ and metal ion ‘C’ increases, which weakens the coordination of metal ion ‘C’ to the dCTP O3γ atom and stimulates substitution of this atom by Glu802:Oε1. In the R696W system, the distance between the catalytic ion ‘A’ and nucleotide-binding ion ‘B’ decreases as the ions move with the fingers away from the primer terminus.
As shown in Fig. 10, in the WT0 system that lacks the incoming nucleotide and ions in the active-site pocket, the catalytic residues move apart from each other and several new interactions form to other protein residues. In addition to the salt-bridge “gates” that Asp764 and Glu802 form with residues in the fingers described above in “Open state enables support of the primer terminus by both the fingers and palm”, Asp608 forms a hydrogen bond to Lys803 of the palm (see orientation of these residues in Fig. S5 and distance data in Fig. S16 in the Supporting Information). Similarly, the absence of metal ion ‘C’ in the WT-2MG system results in Glu802 forming an interaction with Lys678 of the fingers described above and Asp608 forming a hydrogen bond to Lys803 (Fig. 10 and Fig. S16 in the Supporting Information).
Discussion
Elucidating the atomic-level details of pol δ’s mechanism for nucleotide insertion allows the factors that affect its function and fidelity to be identified. Analysis of mutant systems with altered functional properties such as the R696W and A699Q systems allow additional insights to be garnered regarding error-prone replication. Our simulations of several pol δ systems capture many changes occurring in the protein and DNA as summarized in Table 2. The combined results emphasize that subtle system changes can lead to a myriad of conformational adjustments that affect the overall function and fidelity of pol δ.
A chemically inactive pol δ “open” conformation was identified from simulations of the WT0 system, which lacks a dNTP and active-site ions. In this conformation, the fingers are positioned away from both the DNA and exonuclease domain to open up the active site while the α-helix O of the fingers shifts toward the palm, forming a series of salt-bridge “gates” to anchor the primer terminus to the palm and fingers. The repositioning of the fingers occurs when a salt bridge between Arg696 and Asp680 that connects α-helices O and P of the fingers breaks. This movement in the fingers disrupts interactions with the exonuclease domain, beginning with adjacent α-helices F and G, and, consequently, destabilizing the β-hairpin. The thumb tip shifts along the DNA away from the active site to further release restrictions on the active site conformation. All catalytic residues form new interactions with the fingers and palm in the absence of the ions in the active site. Together, these changes produce a relaxed protein/DNA complex that is not poised for a chemical reaction.
The closed conformation, modeled by the WT-3MG system, has a tightly organized active-site geometry with close proximity of the correct incoming nucleotide to the primer terminus and little variation in the positions of the three bound magnesium ions and coordinating active-site residues. These factors suggest the closed form creates a chemically competent state in agreement with the conformational state adopted in the pol δ X-ray crystal structure [26]. A comparison of open and closed pol δ fingers orientations shown in Fig. 3b reveals a significant angle of rotation, but one that is smaller compared to structural data of pol α and RB69 pol [25, 33].
The WT-3CA and WT-2MG systems, which differ only in the species of ion and number of ions from the WT-3MG system, do not appear as well assembled for chemistry. Both the WT-3CA and WT-2MG systems show changes to the fingers and the WT-2MG system has increased fingers-palm residue interactions, similar to what occurs in the open state. In the WT-3CA system, movement in the fingers is associated with transient repositioning in metal ion ‘C’ to disrupt active-site ion coordination. This temporary active site disassembly helps to explain the linkage between calcium ions and pol δ inactivity [26]. In the WT-2MG system, a lengthening in the catalytic distance between O3′ and Pα atoms occurs, which indicates a less-active chemical state that is in agreement with decreased catalytic efficiency data when metal ion ‘C’ is not bound [26].
An open conformation is also adopted by the R696W pol δ mutant, which has reduced polymerase activity [26]. During the simulation, the fingers rotate to the open conformation and hydrogen bonding occurs between the fingers and palm. Loss of the salt bridge between the fingers helices as a result of the Arg696 mutation to tryptophan facilitates the rearrangement of the fingers. This open conformation of the R696W pol δ mutant signals the transition to a chemically inactive state, since the dCTP and ions move with the fingers away from the primer terminus and the catalytic O3′-Pα distance substantially elongates.
Surprisingly, simulations of both the WT-2MG and R696W systems capture a shifting of the thumb away from the DNA that facilitates shifting of the DNA primer strand. Loosening of thumb/DNA interactions facilitates changes in DNA position associated with polymerase cycling as well as switching of the primer strand to the exonuclease domain for excision of incorrect nucleotides. Interestingly, the same thumb region shifts in RB69 pol as seen from a comparison of apo and exonuclease complexes [57]. If pol δ functions similarly to RB69 pol, this thumb motion may initiate the transfer of the DNA primer strand to the exonuclease domain for proofreading. In the RB69 pol system, the thumb shift produces a more open conformation that is proposed to allow transfer of the DNA into the exonuclease active site. Supportingly, the primer terminus base pair frays in the WT-2MG system and the other conformational changes occurring in this system indicate a less active polymerization state. Since the loss of metal ion ‘C’ triggers these changes in the simulation of the WT-2MG system, our results suggest that dissociation of metal ion ‘C’ may occur prior to switching of the DNA primer strand to the exonuclease domain for proofreading. Our simulations of the R696W pol δ mutant support a connection between metal ion ‘C’ and DNA switching since this ion moves further out of the active site as similar thumb and DNA changes occur.
Analysis of the residues in the thumb hinge region between the thumb tip and base suggests cross-interaction that could impact thumb position. In the pol δ thumb hinge, hydrophobic residues such as Leu845 and Leu887 (Fig. S8 in the Supporting Information) are closely packed together. In related enzymes, T4 pol and RB69 pol, the analogous residue to Leu845 in the thumb base is a smaller alanine residue. In T4 pol, mutation of this alanine residue to valine causes an increase in polymerase fidelity [88]. The same effect is proposed for mutation in RB69 since the bulkier residue would favor the thumb conformation adopted by exonuclease complexes and thus would promote proofreading while hampering polymerization activity [25]. Although the leucine residue in pol δ is larger than both alanine and valine, mutation of this residue to an even larger residue such as tryptophan would open the hinge and promote shifting of the thumb tip away from the primer terminus; in a similar fashion, this would promote exonuclease complex formation and increased proofreading activity.
Our simulation results suggest that a conserved exonuclease domain β-hairpin has an active role in pol δ’s proofreading ability and fidelity. In the event that a mismatch was inserted by pol δ, the primer strand would unwind from the template strand and move to the exonuclease active site, where the incorrect nucleotide would be removed by the polymerase’s 3′-5′ exonuclease activity. In our simulations, repositioning of the β-hairpin is stimulated by changes to the active site organization that render it less ready for the chemical reaction. This is supported by the movement of the β-hairpin in the system without the incoming nucleotide, the WT-2MG system that lacks the third ion, and the R696W mutant system when the fingers open and the active site disassembles. The β-hairpin shows no substantial movement in wild-type systems with the correct nucleotide and three metal ions bound to the active site, which have well-organized active sites. Differently, in the A699Q system, β-hairpin movement results from the mutant Gln699 residue disrupting interactions between Tyr446 and Arg470, which help secure the β-hairpin position.
Our simulations reveal that specific β-hairpin residues interact with the DNA that can initiate DNA unwinding. Movement in the β-hairpin allows Tyr446 to come in close contact with the primer terminus and may serve as a “sensor” of active-site conditions needed to determine whether the DNA primer should switch to the exonuclease active site. Once Tyr446 has performed its function, it adopts more energetically stable interactions with the exonuclease domain and movement of the β-hairpin halts. The new orientation of the β-hairpin in the WT-2MG system enables Lys444 to “hold” onto the template base opposite the primer terminus and this starts to break apart that base pair, revealing a mechanism for unwinding the primer strand from the template so that it can enter the exonuclease active site. These β-hairpin/template strand interactions would keep the DNA associated with the polymerase while the primer is shuttled from the polymerase to the exonuclease active site. This proposed function of the β-hairpin is supported by studies analyzing the analogous β-hairpin in RB69 pol [64].
Interestingly, yeast pol δ’s Tyr446 in the β-hairpin loop is conserved in T4 pol (Tyr254) and RB69 pol (Tyr257). At this position in human pol δ there is a threonine residue (Thr441), which retains the terminal hydroxyl group of the tyrosine side chain. Mutation of T4 pol’s Tyr254 brings about significantly more replication errors [65], which supports a role for the β-hairpin in the proofreading process. In addition, the flexibility in β-hairpin position that facilitates changing interactions between Tyr446 and the DNA captured in our pol δ simulations is mirrored in several studies of RB69 pol. A structural study of the exonuclease complex of RB69 pol with DNA containing a thymine glycol lesion shows the β-hairpin positioned between the separated template and primer strands with Tyr257 stacking with an exposed primer-strand base to help stabilize the position of the primer in the exonuclease active site [67]. Other RB69 pol editing complex structures with nonlesioned DNA [57] and with DNA containing an abasic lesion [89] show different specific interactions between the β-hairpin and the separated template and primer strands, suggesting that these interactions may vary depending on the replication context.
The rearrangements occurring in the R696W and A699Q pol δ mutant systems provide mechanisms for the altered function and fidelity profiles of these enzymes. Our simulation results suggest that the loss of the Arg696 salt bridge interactions to the fingers and NTD are the source of the changes occurring in the R696W mutant. In particular, loss of the Arg696 (α-helix P)–Asp680 (α-helix O) occurs in all systems where the fingers exhibit movement, suggesting that mutation in either of these residues would impact polymerase opening-closing movement. Since opening of the fingers signals a transition to an inactive chemical state, this change explains the reduced polymerase activity of the R696W mutant. Continued nucleotide insertion would warrant fingers closing as well as the thumb tip moving back to the DNA primer. Any possible insertion from the open state would be less selective for the correct nucleotide, which is another feature of the R696W mutant. Mismatch complexes of other polymerases in partially open complexes have already suggested how incorrect nucleotides could be accommodated in the active site [90, 91]. Although movement of the thumb away from the DNA implies a transition from polymerase to editing modes, this may not easily occur in the R696W mutant since the orientations of the β-hairpin prevent Tyr446 from forming interactions with the primer terminus and Lys444 from forming interactions with template strand bases. Therefore, the R696W enzyme may have a problem transferring the primer strand to the exonuclease active site for proofreading despite the thumb loosening interactions with the DNA primer. Adding to the inherent fidelity problems of the R696W mutant, it has also been hypothesized that a lowered polymerase activity stimulates the production of dNTPs in cells and that elevated dNTP pool levels promote further DNA synthesis and extension of mismatched primer termini by this mutant enzyme [74].
A different scenario emerges from the A699Q pol δ mutant simulations to explain the mutant’s lowered fidelity. The flexibility of the Gln699 side chain destabilizes the position of the exonuclease domain β-hairpin, allowing the β-hairpin to form even more energetically favorable interactions within the exonuclease domain than occur in the WT-3MG system. This strong interaction prevents the β-hairpin from shifting. We propose that limited mobility in the β-hairpin would result in an impaired ability to check for mismatches and would hamper proofreading, allowing some insertion errors to go undetected. Unlike the R696W system, the closed conformation of the A699Q pol δ mutant is bolstered by interactions between Gln699 and residues in adjacent α-helix J of the NTD. We propose that increased support for the closed fingers conformation would promote nucleotide insertion, potentially even of mismatches. In agreement with the importance of the packing interaction between Met540 and Gln699 for strengthening the closed fingers position when a dNTP is bound, experimental studies of the M540A/A699Q double mutant show that the enzyme’s fidelity is similar to that of wild-type pol δ [76]. This suggests that removal of the interaction between Met540 and Gln699 allows the fingers to move between open and closed conformations as readily as the wild-type polymerase, which allows for greater nucleotide selectivity. In addition, studies of Y537A and the Y537A/A699Q double mutant suggest that Tyr537 has a stabilizing role in the polymerase complex that could be substituted by Gln699 and not Ala699 [76]. This agrees with our simulation results, indicating a supportive role of Tyr537 in maintaining the closed fingers conformation. In sum, these factors involving the β-hairpin and strengthened closed conformation would facilitate increased numbers of base substitutions and other errors occurring during replication, and explain the lowered fidelity of the A699Q pol δ mutant.
Summary
The combined results of our six pol δ simulations, including two mutant forms, provide evidence for multiple conformational changes and specific interactions that contribute to the function and fidelity of pol δ. In particular, we find that a rotation in the fingers transitions pol δ from an open- to a closed-conformational state to produce a well-organized active site when the correct nucleotide and three magnesium ions are bound. The open state also exhibits close ties between the fingers and palm to secure the primer terminus to the active site. Loss of metal ion ‘C’ disrupts the assembly of the active site and stimulates conformational changes in the thumb and a conserved β-hairpin in the exonuclease domain that facilitate movement in the DNA primer strand and breakage of the primer terminus base pair. These rearrangements suggest a mechanism involving Lys444 and Tyr446 of the β-hairpin for unwinding the primer strand from the template strand to transition the primer strand to the exonuclease domain for editing. Our simulations reveal that any change to the active site (i.e., in divalent ions and the dNTP) or mutations in the fingers, bring about conformational changes in the β-hairpin. In both mutant systems, disruption of the open- to closed-conformational change process affects proper nucleotide selection. In the A699Q system, interactions involving mutated residue Gln699 with the NTD and exonuclease domain serve to support the closed conformation and repositioning of the β-hairpin away from the DNA implies reduced proofreading ability. Differently, the R696W pol δ mutant transitions to an open conformational state while the correct nucleotide and ions are bound to account for reduced polymerase activity and lowered nucleotide selectivity. In sum, a delicate balance of domain motions and specific residue interactions account for the proper functioning and high fidelity of pol δ.
References
Goffeau A, Barrell BG, Bussey H et al (1996) Life with 6000 genes. Science 274:546–567. https://doi.org/10.1126/science.274.5287.546
Venter JC, Adams MD, Myers EW et al (2001) The sequence of the human genome. Science 291:1304–1351. https://doi.org/10.1126/science.1058040
Lander ES, Linton LM, Birren B et al (2001) Initial sequencing and analysis of the human genome. Nature 409:860–921. https://doi.org/10.1038/35057062
Bebenek K, Kunkel TA (2004) Functions of DNA polymerases. Adv Protein Chem 69:137–165
Garg P, Burgers PM (2005) DNA polymerases that propagate the eukaryotic DNA replication fork. Crit Rev Biochem Mol Biol 40:115–128
Nick McElhinny SA, Gordenin DA, Stith CM et al (2008) Division of labor at the eukaryotic replication fork. Mol Cell 30:137–144
Pursell ZF, Isoz I, Lundström E-B et al (2007) Yeast DNA polymerase ε participates in leading-strand DNA replication. Science 317:127–130. https://doi.org/10.1126/science.1144067
Clausen AR, Lujan SA, Burkholder AB et al (2015) Tracking replication enzymology in vivo by genome-wide mapping of ribonucleotide incorporation. Nat Struct Mol Biol 22:185–191. https://doi.org/10.1038/nsmb.2957
Daigaku Y, Keszthelyi A, Müller CA et al (2015) A global profile of replicative polymerase usage. Nat Struct Mol Biol 22:192–198. https://doi.org/10.1038/nsmb.2962
Johnson RE, Klassen R, Prakash L, Prakash S (2015) A major role of DNA polymerase δ in replication of both the leading and lagging DNA strands. Mol Cell 59:163–175
Reijns MAM, Kemp H, Ding J et al (2015) Lagging-strand replication shapes the mutational landscape of the genome. Nature 518:502–506. https://doi.org/10.1038/nature14183
Miyabe I, Mizuno K, Keszthelyi A et al (2015) Polymerase δ replicates both strands after homologous recombination-dependent fork restart. Nat Struct Mol Biol 22:932–938. https://doi.org/10.1038/nsmb.3100
Yeeles JTP, Janska A, Early A, Diffley JFX (2017) How the eukaryotic replisome achieves rapid and efficient DNA replication. Mol Cell 65:105–116. https://doi.org/10.1016/j.molcel.2016.11.017
Prindle MJ, Loeb LA (2012) DNA polymerase delta in DNA replication and genome maintenance. Environ Mol Mutagen 53:666–682. https://doi.org/10.1002/em.21745
Dilley RL, Verma P, Cho NW et al (2016) Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 539:54–58. https://doi.org/10.1038/nature20099
McCulloch SD, Kunkel TA (2008) The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res 18:148–161. https://doi.org/10.1038/cr.2008.4
St Charles JA, Liberti SE, Williams JS et al (2015) Quantifying the contributions of base selectivity, proofreading and mismatch repair to nuclear DNA replication in Saccharomyces cerevisiae. DNA Repair 31:41–51. https://doi.org/10.1016/j.dnarep.2015.04.006
Fortune JM, Pavlov YI, Welch CM et al (2005) Saccharomyces cerevisiae DNA polymerase δ high fidelity for base substitutions but lower fidelity for single-and multi-base deletions. J Biol Chem 280:29980–29987
Schmitt MW, Matsumoto Y, Loeb LA (2009) High fidelity and lesion bypass capability of human DNA polymerase δ. Biochimie 91:1163–1172
Pavlov YI, Frahm C, Nick McElhinny SA et al (2006) Evidence that errors made by DNA polymerase alpha are corrected by DNA polymerase delta. Curr Biol CB 16:202–207. https://doi.org/10.1016/j.cub.2005.12.002
Flood CL, Rodriguez GP, Bao G et al (2015) Replicative DNA polymerase δ but not ε proofreads errors in cis and in trans. PLoS Genet 11:e1005049. https://doi.org/10.1371/journal.pgen.1005049
Steitz TA (1999) DNA polymerases: structural diversity and common mechanisms. J Biol Chem 274:17395–17398
Braithwaite DK, Ito J (1993) Compilation, alignment, and phylogenetic relationships of DNA polymerases. Nucleic Acids Res 21:787–802
Wang J, Satter AKMA, Wang CC et al (1997) Crystal structure of a pol α family replication DNA polymerase from bacteriophage RB69. Cell 89:1087–1099
Franklin MC, Wang J, Steitz TA (2001) Structure of the replicating complex of a pol alpha family DNA polymerase. Cell 105:657–667
Swan MK, Johnson RE, Prakash L et al (2009) Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase δ. Nat Struct Mol Biol 16:979–986
Wang F, Yang W (2009) Structural insight into translesion synthesis by DNA Pol II. Cell 139:1279–1289. https://doi.org/10.1016/j.cell.2009.11.043
Perera RL, Torella R, Klinge S et al (2013) Mechanism for priming DNA synthesis by yeast DNA polymerase α. Elife 2:e00482
Jain R, Rajashankar KR, Buku A et al (2014) Crystal structure of yeast DNA polymerase epsilon catalytic domain. PLoS One 9(4):e94835
Hogg M, Osterman P, Bylund GO et al (2014) Structural basis for processive DNA synthesis by yeast DNA polymerase ɛ. Nat Struct Mol Biol 21:49–55
Doublié S, Zahn KE (2014) Structural insights into eukaryotic DNA replication. Front Microbiol 5:444
Baranovskiy AG, Babayeva ND, Suwa Y et al (2014) Structural basis for inhibition of DNA replication by aphidicolin. Nucleic Acids Res 42:14013–14021. https://doi.org/10.1093/nar/gku1209
Coloma J, Johnson RE, Prakash L et al (2016) Human DNA polymerase alpha in binary complex with a DNA: DNA template-primer. Sci Rep 6:23784. https://doi.org/10.1038/srep23784
Johnson KA (2008) Role of induced fit in enzyme specificity: a molecular forward/reverse switch. J Biol Chem 283:26297–26301. https://doi.org/10.1074/jbc.r800034200
Ahn J, Werneburg BG, Tsai M-D (1997) DNA polymerase β: structure-fidelity relationship from pre-steady-state kinetic analyses of all possible correct and incorrect base pairs for wild type and R283A mutant. Biochemistry 36:1100–1107
Sawaya MR, Prasad R, Wilson SH et al (1997) Crystal structures of human DNA polymerase β complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry 36:11205–11215
Wong I, Patel SS, Johnson KA (1991) An induced-fit kinetic mechanism for DNA replication fidelity: direct measurement by single-turnover kinetics. Biochemistry 30:526–537
Doublié S, Tabor S, Long AM et al (1998) Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature 391:251–258. https://doi.org/10.1038/34593
Dahlberg ME, Benkovic SJ (1991) Kinetic mechanism of DNA polymerase I (Klenow fragment): identification of a second conformational change and evaluation of the internal equilibrium constant. Biochemistry 30:4835–4843
Li Y, Korolev S, Waksman G (1998) Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J 17:7514–7525
Suo Z, Johnson KA (1998) Selective inhibition of HIV-1 reverse transcriptase by an antiviral inhibitor, (R)-9-(2-Phosphonylmethoxypropyl)adenine. J Biol Chem 273:27250–27258
Huang H, Chopra R, Verdine GL, Harrison SC (1998) Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 282:1669–1675
Arora K, Schlick T (2004) In silico evidence for DNA polymerase β’s substrate-induced conformational change. Biophys J 87:3088–3099
Arora K, Beard WA, Wilson SH, Schlick T (2005) Mismatch-induced conformational distortions in polymerase β support an induced-fit mechanism for fidelity. Biochemistry 44:13328–13341
Radhakrishnan R, Schlick T (2004) Orchestration of cooperative events in DNA synthesis and repair mechanism unravelled by transition path sampling of DNA polymerase β’s closing. Proc Natl Acad Sci USA 101:5970–5975
Florián J, Warshel A, Goodman MF (2002) Molecular dynamics free-energy simulations of the binding contribution to the fidelity of T7 DNA polymerase. J Phys Chem B 106:5754–5760. https://doi.org/10.1021/jp020791m
Miller BR, Parish CA, Wu EY (2014) Molecular dynamics study of the opening mechanism for DNA polymerase I. PLoS Comput Biol 10:1003961. https://doi.org/10.1371/journal.pcbi.1003961
Miller BR, Beese LS, Parish CA, Wu EY (2015) The closing mechanism of DNA polymerase I at atomic resolution. Structure 23:1609–1620. https://doi.org/10.1016/j.str.2015.06.016
Meli M, Sustarsic M, Craggs TD et al (2016) DNA polymerase conformational dynamics and the role of fidelity-conferring residues: insights from computational simulations. Front Mol Biosci 3:20. https://doi.org/10.3389/fmolb.2016.00020
Kirmizialtin S, Nguyen V, Johnson KA, Elber R (2012) How conformational dynamics of DNA polymerase select correct substrates: experiments and simulations. Structure 20:618–627
Kirmizialtin S, Johnson KA, Elber R (2015) Enzyme selectivity of HIV reverse transcriptase: conformations, ligands, and free energy partition. J Phys Chem B 119:11513–11526. https://doi.org/10.1021/acs.jpcb.5b05467
Wang J, Sattar AK, Wang CC et al (1997) Crystal structure of a pol alpha family replication DNA polymerase from bacteriophage RB69. Cell 89:1087–1099
Garcia-Diaz M, Bebenek K, Krahn JM et al (2005) A closed conformation for the pol λ catalytic cycle. Nat Struct Mol Biol 12:97–98
Foley MC, Arora K, Schlick T (2006) Sequential side-chain residue motions transform the binary into the ternary state of DNA polymerase λ. Biophys J 91:3182–3195
Moon AF, Pryor JM, Ramsden DA et al (2014) Sustained active site rigidity during synthesis by human DNA polymerase mu. Nat Struct Mol Biol 21:253–260. https://doi.org/10.1038/nsmb.2766
Li Y, Schlick T (2010) Modeling DNA polymerase mu motions: subtle transitions before chemistry. Biophys J 99:3463–3472. https://doi.org/10.1016/j.bpj.2010.09.056
Shamoo Y, Steitz TA (1999) Building a replisome from interacting pieces: sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex. Cell 99:155–166
Kropp HM, Betz K, Wirth J et al (2017) Crystal structures of ternary complexes of archaeal B-family DNA polymerases. PLoS One 12:0188005. https://doi.org/10.1371/journal.pone.0188005
Nakamura T, Zhao Y, Yamagata Y et al (2012) Watching DNA polymerase η make a phosphodiester bond. Nature 487:196–201. https://doi.org/10.1038/nature11181
Gao Y, Yang W (2016) Capture of a third Mg2+ is essential for catalyzing DNA synthesis. Science 352:1334–1337. https://doi.org/10.1126/science.aad9633
Freudenthal BD, Beard WA, Shock DD, Wilson SH (2013) Observing a DNA polymerase choose right from wrong. Cell 154:157–168. https://doi.org/10.1016/j.cell.2013.05.048
Freudenthal BD, Beard WA, Perera L et al (2015) Uncovering the polymerase-induced cytotoxicity of an oxidized nucleotide. Nature 517:635–639. https://doi.org/10.1038/nature13886
Marquez LA, Reha-Krantz LJ (1996) Using 2-aminopurine fluorescence and mutational analysis to demonstrate an active role of bacteriophage T4 DNA polymerase in strand separation required for 3′ --> 5′-exonuclease activity. J Biol Chem 271:28903–28911
Hogg M, Aller P, Konigsberg W et al (2007) Structural and biochemical investigation of the role in proofreading of a beta hairpin loop found in the exonuclease domain of a replicative DNA polymerase of the B family. J Biol Chem 282:1432–1444. https://doi.org/10.1074/jbc.M605675200
Subuddhi U, Hogg M, Reha-Krantz LJ (2008) Use of 2-aminopurine fluorescence to study the role of the beta hairpin in the proofreading pathway catalyzed by the phage T4 and RB69 DNA polymerases. Biochemistry 47:6130–6137. https://doi.org/10.1021/bi800211f
Trzemecka A, Płochocka D, Bebenek A (2009) Different behaviors in vivo of mutations in the beta hairpin loop of the DNA polymerases of the closely related phages T4 and RB69. J Mol Biol 389:797–807. https://doi.org/10.1016/j.jmb.2009.04.055
Aller P, Duclos S, Wallace SS, Doublié S (2011) A crystallographic study of the role of sequence context in thymine glycol bypass by a replicative DNA polymerase serendipitously sheds light on the exonuclease complex. J Mol Biol 412:22–34. https://doi.org/10.1016/j.jmb.2011.07.007
Hadjimarcou MI, Kokoska RJ, Petes TD, Reha-Krantz LJ (2001) Identification of a mutant DNA polymerase delta in Saccharomyces cerevisiae with an antimutator phenotype for frameshift mutations. Genetics 158:177–186
Darmawan H, Harrison M, Reha-Krantz LJ (2015) DNA polymerase 3′→5′ exonuclease activity: different roles of the beta hairpin structure in family-B DNA polymerases. DNA Repair 29:36–46. https://doi.org/10.1016/j.dnarep.2015.02.014
Ganai RA, Bylund GO, Johansson E (2015) Switching between polymerase and exonuclease sites in DNA polymerase ε. Nucleic Acids Res 43:932–942. https://doi.org/10.1093/nar/gku1353
Palles C, Cazier J-B, Howarth KM et al (2013) Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 45:136–144
Briggs S, Tomlinson I (2013) Germline and somatic polymerase ε and δ mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol 230:148–153. https://doi.org/10.1002/path.4185
Flohr T, Dai J, Büttner J et al (1999) Detection of mutations in the DNA polymerase δ gene of human sporadic colorectal cancers and colon cancer cell lines. Int J Cancer 80:919–929
Mertz TM, Sharma S, Chabes A, Shcherbakova PV (2015) Colon cancer-associated mutator DNA polymerase δ variant causes expansion of dNTP pools increasing its own infidelity. Proc Natl Acad Sci USA 112:E2467–E2476
Daee DL, Mertz TM, Shcherbakova PV (2010) A cancer-associated DNA polymerase δ variant modeled in yeast causes a catastrophic increase in genomic instability. Proc Natl Acad Sci USA 107:157–162
Prindle MJ, Schmitt MW, Parmeggiani F, Loeb LA (2013) A substitution in the fingers domain of DNA polymerase δ reduces fidelity by altering nucleotide discrimination in the catalytic site. J Biol Chem 288:5572–5580
Lee EH, Hsin J, Sotomayor M et al (2009) Discovery through the computational microscope. Structure 17:1295–1306. https://doi.org/10.1016/j.str.2009.09.001
Dror RO, Dirks RM, Grossman JP et al (2012) Biomolecular simulation: a computational microscope for molecular biology. Annu Rev Biophys 41:429–452. https://doi.org/10.1146/annurev-biophys-042910-155245
Brooks BR, Brooks III CL, MacKerell JAD et al (2009) CHARMM: the biomolecular simulation program. J Comput Chem 30:1545–1614
Brooks BR, Bruccoleri RE, Olafson BD et al (1983) CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 4:187–217
Humphrey W, Dalke A, Schulten K (1996) VMD-visual molecular dynamics. J Mol Graph 14:33–38
Phillips JC, Braun R, Wang W et al (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802
MacKerell AD Jr, Bashford D, Bellott M, et al. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102:3586–3616
MacKerell AD Jr, Banavali NK (2000) All-atom empirical force field for nucleic acids: II. Application to molecular dynamics simulations of DNA and RNA in solution. J Comput Chem 21:105–120
Feller SE, Zhang Y, Pastor RW, Brooks BR (1995) Constant pressure molecular dynamics simulation: the Langevin piston method. J Chem Phys 103:4613–4621
Ryckaert J-P, Ciccotti G, Berendsen HJC (1977) Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23:327–341
Darden TA, York DM, Pedersen LG (1993) Particle mesh Ewald: an N*log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092
Reha-Krantz LJ (1989) Locations of amino acid substitutions in bacteriophage T4 tsL56 DNA polymerase predict an N-terminal exonuclease domain. J Virol 63:4762–4766
Hogg M, Wallace SS, Doublié S (2004) Crystallographic snapshots of a replicative DNA polymerase encountering an abasic site. EMBO J 23:1483–1493. https://doi.org/10.1038/sj.emboj.7600150
Wu EY, Beese LS (2011) The structure of a high fidelity DNA polymerase bound to a mismatched nucleotide reveals an “ajar” intermediate conformation in the nucleotide selection mechanism. J Biol Chem 286:19758–19767. https://doi.org/10.1074/jbc.m110.191130
Benitez BAS, Arora K, Balistreri L, Schlick T (2008) Mismatched base-pair simulations for ASFV pol X/DNA complexes help interpret frequent G•G misincorporation. J Mol Biol 384:1086–1097
Acknowledgments
This research utilized resources at the New York Center for Computational Sciences at Stony Brook University/Brookhaven National Laboratory which is supported by the United States Department of Energy under Contract No. DE-AC02-98CH10886 and by the State of New York. In addition, this research used resources from the Rutgers Discovery Informatics Institute (RDI2), New Jersey’s Center for Advanced Computation, a user facility supported by the Rutgers University Office of Research and Economic Development (ORED). We thank New York City high school students K. Alvarez and S. Woolward supported by the American Chemical Society (ACS) Project Seed for their efforts with the initial data analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 12104 kb)
Rights and permissions
About this article

Cite this article
Foley, M.C., Couto, L., Rauf, S. et al. Insights into DNA polymerase δ’s mechanism for accurate DNA replication. J Mol Model 25, 80 (2019). https://doi.org/10.1007/s00894-019-3957-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-3957-z