
Abstract
The scientific community has shown particular interest in the study of quinolinones—a class of bicyclic organic compounds. An example of these compounds are the 4-quinolinones, considered to be very useful building blocks, since they can adapt their molecular structures with different ligands for applications in various fields such as pharmacy, medicine, physics and engineering. The compounds (E)-3-(benzylidene)-2-(3-nitrophenyl)-2,3-dihydro-1-(phenylsulfonyl)-quinolin-4-(1H)-one (NFQ) and (E)-3-(benzylidene)-2-(4-bromophenyl)-2,3-dihydro-1-(phenylsulfonyl) quinolin-4-(1H)-one (BFQ) were synthesized and characterized by infrared spectroscopy, 1H and 13C NMR, and melting point. NFQ crystallized in the orthorhombic Pbca space group while BFQ appears in the monoclinic P21/n space group. X-ray diffraction was used to evaluate their crystallographic structures, and Hirshfeld surface evaluates the intermolecular interactions, supramolecular arrangement and packaging. Theoretical vibrational assignments and calculated electronic properties also demonstrate acceptable agreement between experimental and theoretical results.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Quinolinones are a class of organic compounds known for their antimicrobial activity. Quinolinones are efficient in the treatment of infection by Gram-positive and Gram-negative bacteria, and are used widely in human and veterinary medicine [1]. There is considerable scientific interest in these substances, which has been increasing steadily since their discovery more than 50 years ago, when nalidixic acid was created accidentally in 1962 by Lesher et al. [2, 3]. These compounds have been used widely for the development of new drugs, since they have an ideal structure for application [4]. This fact is confirmed by the large number of patents registered recently [5,6,7,8], as well as the many studies of their biological potential [9,10,11,12,13,14,15], including use as an antitumor agent [16, 17] and in engineering [18]. Studies have shown that quinolinones with different binders can serve as a “building block of choice” [19] because they allow the entry of different binders in various positions, justifying a wide range of applications.
However, for a complete understanding of new structures, detailed studies are necessary to obtain a more accurate idea of structure–activity relationships. Structural insights into the crystalline solid state, in addition to a thorough understanding of the atomic architecture of the molecular conformation, have transformed industries and created new frontiers, including the development of new drugs and materials [20]. Therefore, to understand further the solid arrangement of two new 4-quinolinones, this work proposes the acquisition, structural elucidation and quantum theoretical calculations of (E)-3-(benzylidene)-2-(3-nitrophenyl)-2,3-dihydro-1-(phenylsulfonyl)-quinolin-4-(1H)-one (NFQ) and (E)-3-(benzylidene)-2-(4-bromophenyl)-2,3-dihydro-1-(phenylsulfonyl) quinolin-4-(1H)-one (BFQ). For this purpose, the packing, crystalline arrangement, angular analysis and interatomic distances, molecular geometry, intermolecular interactions, Hirshfeld surface (HS), fingerprints, theoretical vibrational assignments and electronic properties of NFQ and BFQ compounds were elucidated.
Experimental and computational procedures
Synthesis and crystallization
NFQ (4) and BFQ (5) were obtained from sulfonamide chalcones (1, 2) and reacted with benzaldehyde (3). The resulting precipitate (4, 5) was purified by slow recrystallization from dichloromethane (CH2Cl2) and ethanol (CH3CH2OH) (4:1), after drying. (4) Yield: 43.8%, boiling point: 184–185 °C, 1H MNR (CDCl3, 500 MHz): δ 6.79 (s, 1H), δ 7.13 (m, 2H), δ 7.18 (d, J 7.32 Hz, 2 H), δ 7.21 (m, 2H), δ 7.31 (td, J 7.63 Hz, J 1.22 Hz, 1H), δ 7.43 (m, 2H), δ 7.47 (m, 2H), δ 7.52 (m, 1H), δ 7.57 (td, J 7.78 Hz, J 1.83 Hz, 1H), δ 7.72 (s, 1H), δ 7.77 (dd, J 8.09 Hz, J 0.76 Hz, 1H), δ 7.82 (dt, J 7.93 Hz, J 0.92 Hz, 1H), δ 7.89 (dd, J 7.63 Hz, J 1.53 Hz, 1H), δ 8.12 (ddd, J 8.39 Hz, J 1.07 Hz, J 0.92 Hz, 1H), δ 8.28 (s, 1H); IV (cm−1): 3091 (m), 3057 (m), 1670 (vs), 1604 (vs), 1529 (vs), 1460 (vs), 1180 (vs), 1055(s), 970 (s). (5) Yield: 43.39%, boiling point: 181–183 °C, 1H MNR (CDCl3, 500 MHz) δ 6.65 (s, 1H), δ 7.11 (dd, J 8.55 Hz, 1.22 Hz, 2H), δ 7.20 (m, 2H), δ 7.29 (m, 1H), δ 7.32 (d, J 8.85 Hz, 2H), δ 7.42 (m, 4H), δ 7.45 (m, 1H), δ 7.49 (t, J 7.48 Hz, 1H), δ 7.55 (td, J 7.78 Hz, J 1.53 Hz, 1H), δ 7.63 (s, 1H), δ 7.72 (d, J 8.24 Hz, J 0.61 Hz, 1H), δ 7.89 (dd, J 7.78 Hz, J 1.37 Hz, 1H); IV (cm−1): 1669 (s), 1607 (vs), 1458 (s), 1359 (s), 1301 (s), 1171 (s), 761 (s) (Scheme 1).

Synthesis of (E)-3-(benzylidene)-2-(3-nitrophenyl)-2,3-dihydro-1-(phenylsulfonyl)-quinolin-4-(1H)-one (NFQ) (4) and (E)-3-(benzylidene)-2-(4-bromophenyl)-2,3-dihydro-1-(phenylsulfonyl) quinolin-4-(1H)-one (BFQ) (5)
Crystallographic characterization
X-Ray diffraction data of NFQ and BFQ were collected at room temperature with a Bruker Apex II CCD diffractometer equipped with graphite monochromated MoKα radiation on λ equal to 0.71073 Å (Bruker, Billerica, MA). Data was processed using Bruker software APEX2 [21] and the crystallographic structures were solved by direct methods, with refinements undertaken using the least square method in SHELX2016 [22] software. Hydrogen atoms were then placed in calculated positions and refined with fixed individual displacement parameters [Uiso(H) = 1.2Ueq or 1.5Ueq] according to the riding model (C–H bond lengths of 0.97 Å and 0.96 Å for aromatic and methyl groups, respectively). The software WinGX [23], Ortep [23] and Mercury [24] was employed to obtain the structural information presented here as tables, figures and molecular representations. The PLATON [25] program was used to check possible interaction between hydrogen bonds.
Hirshfeld surface
To evaluate intermolecular interactions in the crystal packing of NFQ and BFQ, Hirshfeld surface (HS) analysis was used, with Crystal Explorer software [26, 27]. For both molecules, we generated the dnorm and shape index surfaces in addition to a fingerprint plot. These graphical representations allow a better understanding of the interactions using different scales throughout the surface [28], thus presenting different topologies depending on the atom type and the nature of molecular interactions in the compound [29]. The basic idea in creating an HS is to divide the crystal into portions where the electron distribution (ρ) of molecule dominates the electron distribution of the entire crystal. This is called the weighting function w(r), and, in mathematical terms, can be expressed by [27]:
When the aim is to analyze the nature and presence of close contacts, the distance function is very useful. An acceptor and a donor region of intermolecular contact can be indicated as normalized contact distances (dnorm). This surface is composed of two regions, both of which take atoms as a reference; the di region is defined by the distance between an atom within the HS up to its top, while the de region is defined by the distance from an atom that is outside the HS to the HS. In mathematical terms, dnorm is given by [30]:
Besides that, hydrophobic interactions can be recognized using HS through Shape Index analyses. Moreover, when di and de were combined in a two-dimensional (2D) graph (fingerprint), a complete quantitative statistical description is obtained on the occurrence of each type of interaction for a given compound, and this plot is singular for each structure analysed [27].
Theoretical calculations
The geometric parameters of NFQ and BFQ were submitted to theoretical calculations after being solved and refined by crystallographic means, and the resulting CIF file was converted into an input file for Gaussian 09 routine. The starting geometry was fully optimized using density functional theory (DFT) with M06-2X functional [31] and 6–311 + G(d,p) as basis set. In the results, a scaling factor of 0.940 [32] was applied to account for the values of vibrational frequencies [33, 34], after VEDA 4 [35] and Gaussview [36] software were used to assign the vibrational modes.
Results and discussion
Solid state characterization
The hybrid compounds NFQ and BFQ are 1–4 quinolinone derivatives that have a common backbone constituted by a benzenesulfonamide group connected with the nitrogen in para position and a benzylidene in ortho position relative to the ketone group, forming an α–β in saturation with the benzylidene. NFQ possesses an m-nitrobenzyl group connected with C10 and is resolved in the orthorhombic crystal system, spatial group Pbca, and group of Laue mmm. Thus, there is one molecule in each asymmetric unit. For the BFQ compound, which has a p-bromobenzyl group connected with C10, the monoclinic crystalline system, spatial group P21/n and Laue 2/m group were observed. In this case, a molecule was also observed in the asymmetric unit, but there were only four asymmetric units in the unit cell. Data relating to the refinement of the analyzed compound, such as chemical information, correction methods, crystallographic information and other details, are listed in Table 1.
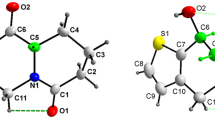
The phenylsulfonyl group is at a distance of 1.659 Å from S1 in NFQ and by 1.661 Å in BFQ, a C10 linked m-nitrophenyl group at a distance of 1.529 Å from C17 in NFQ and a C10-linked para-bromophenyl group 1.527 Å from C17 in BFQ, which are quite similar distances for both molecules. Since C10 is a stereocenter and all compounds crystallized in centrosymmetric space groups, all crystals are composed of racemates, and the asymmetric units presented in Fig. 1 are only R enantiomers. Ortep representation illustrating the structure obtained via X-ray diffraction of NFQ and BFQ is presented in Fig. 1.

ORTEP representation of the (E)-3-(benzylidene)-2-(3-nitrophenyl)-2,3-dihydro-1-(phenylsulfonyl)-quinolin-4-(1H)-one (NFQ) molecule on the left and the (E)-3-(benzylidene)-2-(4-bromophenyl)-2,3-dihydro-1-(phenylsulfonyl) quinolin-4-(1H)-one (BFQ) on the right showing the ellipsoids with probability of 50%
When analyzing the geometric parameters, it is apparent that the unsaturated ketone group presents an E-trans conformation, whose hydrogen atom H9 is oriented in the opposite direction to the D ring. The double bond of the [O1-C7] ketone group of 1.221 Å in NFQ and 1.223 Å in BFQ 1.221 Å is a characteristic Csp2–O bond; likewise, the double bond of the nitro [N2–O4] group of 1.217 Å in the NFQ is a characteristic Nsp2–O bond, both confirmed by values significantly smaller than the binding Ssp3–N [S1–N1] of 1.659 Å in the NFQ and 1.661 Å in the BFQ. Also, on the length between the bonds, the presence of the C8 and C9 insertion, a Csp2–Csp2 bond, with 1.335 Å in the NFQ and 1.342 Å in the BFQ, can be confirmed when compared to the Csp3–C [C10–C17] of 1.529 Å in NFQ and 1.527 Å in BFQ.
On binding angles, it is notable that the molecular architecture presents a tetrahedral geometry, and a trigonal plane in different parts of the molecules. As C10 is a sp3 carbon, this geometry can be observed between [C17–C10–N1] with angles equal to 107.1° in NFQ and 107.61° for BFQ, and between [N1–C10–C8] with angles equal to 110.6° in NFQ and 109.88° for BFQ. However, between [C17–C10–C8], a greater angle of 115.2° in NFQ and 114.32° for BFQ is found, which is due to the presence of ring C. This fact recurs between [C10–N1–C2], which presents an angle of 114.6° in NFQ and 115.34° for BFQ due to ring A. Similar data were reported by Silva [37] when describing the flavonoid C16H14O3. Another site where tetrahedral geometry is seen occurs between atoms S1, C23 and N1, which have angular intervals between 105.44° and 108.73° for NFQ, and 105.55° and 108.59° for BFQ. However, in the same way as for C10, S1 in relation to O2 and O3 shows an angle around 120.40° in NFQ and 120.33° for BFQ, which is probably due to ring B and the sulfur sp3 hybridization. Additionally, trigonal or triangular trigonal geometry can be observed in the aromatic rings A, B, C and E, where the flat trigonal geometry dominates, with ab angular range of 117.8° to 123.0° in NFQ, and 115.7° to 126.2° in BFQ, in the nitro group of the NFQ bound in the C-ring meta-position with an angle of 123.2 (2)° between [O4–N2–O5] and on the carbonyl portion involving the C7, C8, C1 and O1 atoms in both NFQ and BFQ.
When NFQ and BFQ are compared with similar structures [38, 39, 40] the only interatomic distance that presented variation was C7–C8 binding, exhibiting a slightly larger distance. While most structures show an interatomic distance of C7–C8 equivalent carbons between 1.45 and 1.47 Å, this distance in compound 13 was 1.492 Å. Of the 80 similar molecules, this distance was found in only two [38, 41], that is, agreement less than 3%. For comparisons of structural conformers for NFQ and BFQ, the overlapping of the two structures was done by fixing aromatic ring E (Fig. 2). The main structural differences were caused by changing from the m-nitro group to the p-bromine group of aromatic C (RMSD = 0.00498). Among the possibilities for these differences are: (1) a slight twist of the benzylidene ring (C8–C9–C11–C12 = 145.48° for NFQ and 146.06° for BFQ); (2) torsion at heterocyclic ring B in the quinolinone backbone (C17–C10–C8–C7 = −82.38° for NFQ and −74.95° for BFQ); (3) another slight twist at the benzyl ring C (N1–C10–C17–C22 = 48.49° for NFQ and 54.97° for BFQ). The angle between the plane formed by aromatic rings E and B is also similar between the two structures: 57.98° for NFQ and 56.14° for BFQ.

Overlapping of BFQ and NFQ, showing the conformational differences between the compounds
No strong intramolecular interactions were found for compounds NFQ and BFQ, with only weak intermolecular interactions occurring, which stabilize the crystalline structure. This type of interaction occurs due to the low electronegativity in the carbon atom and the weak acidity of the donated proton that connects to a strong acceptor, commonly called non-classical interactions [42]. Table 2 presents values of intermolecular interactions and the respective distances between the donor and hydrogen atoms d(D–H), between the hydrogen and the acceptor atom d(H⋯A) (Å) and between the donor atom and acceptor d(D⋯A) (Å), likewise the angle between donor and acceptor atom θ (D-H⋯A) and the symmetry code.
NFQ packing is stabilized by intermolecular interactions characterized by centrosymmetric chains positioned relative to another as illustrated in Fig. 3. Interaction Ϙ1, illustrated in Fig. 3a, type CaroH⋯Aro-π [42], can be observed between ring B and atoms C20–H20 present in the m-nitrophenyl group that forms a chain running along [100]. Another interaction is seen between atoms C15 and H15 of the benzylidene and the acceptor O3 of the nitro group, forming a C(10) motif interaction Ϙ2 that runs along the [100] axis shown in Fig. 3b. The sulfonyl group also presents a chain interaction (Ϙ3) with atoms C12–H12 present in the benzylidene group demonstrated in Fig. 3c. In this case, a C(9) motif is observed growing along [001]. The BFQ molecule involves a dimer where the acceptor atom H18 of ring C and ring B interacts to form a \( {R}_2^2(14) \) motif. The last intermolecular interaction is a centrosymmetric chain observed in the BFQ molecule forming a \( {C}_1^1(8) \) motif running along [110]. Another important aspect is the presence of NO2 and Br groups in the molecules. These groups make the aromatic ring π electron deficient and, although the bromine is weaker than nitro as an electron-withdrawing group, the first one influences in meta position, local to the C18–H18⋯π interaction in which 2.802 Å is positioned. This fact contributes to the smaller interaction on BFQ in comparison to the C20–H20⋯π NFQ interaction, which has a value of 2.85 Å.

Bidimensional projection of three centrosymmetric chains and involved atoms of NFQ molecules in directions a [100], b [100] and c [110]
The intramolecular interactions in NFQ and BFQ are represented in Figs. 4 and 5, respectively. These interactions given by Crystal Explorer 3.1 software [26, 27] are represented thus: the redder the region, the stronger the interaction. The blue and red regions represent electropositive and electronegative regions of the molecule, respectively. It can be seen in the first figure that both the receptor (1) region and the donor region (2) of the Ϙ2 interaction between the C15-–H15···O3 atoms are stronger than the other interactions, which suggests that this interaction makes a more effective contribution to crystalline packaging. The small distance H15···O3 = 2.466 Å was noted to be the smallest among the different interactions corresponding to its significant contribution of crystalline packaging. For the BFQ molecule, the strongest interaction is seen in Fig. 5b, where the receptor region (12) and donor region (13) of the Ϙ2 interaction responsible for dimer formation are the strongest, and also have the smallest distance interaction. The longer distance 2.802 Å shown in regions (14) acceptor and (15) donor in Fig. 5d is responsible for π interactions in BFQ (a weaker interaction, light red). In the same way, for NFQ, it is seen that the Ϙ1 interaction—the only π interaction between the C20–H20⋯π atoms, shown in Fig. 4b by weak donor region (3) and acceptor region (4)—shows the weakest interaction of the three. This can be proved by the distance between the H20 hydrogen and the π-bond of the C-ring of 2.85 Å–the longest distance. Additionally, the donor region (5) and acceptor region (6) of interaction Ϙ3 between C 12–H 12···O5 is illustrated in Fig. 4c.

a Hirshfeld surface (HS) of dnorm, interactions and fingerprints of compound NFQ for visualization of the interatomic contacts from non-classical hydrogen bonds C15–H15⋯O3 with donor region (1) and receptor region (2); b fingerprint interactions O = H; c fingerprint interactions H⋯H; d C20–H20⋯π specifying the donor region (3) and the recipient region (4); (e) C12–H12⋯O5 with donor region (5) and acceptor region (6); f fingerprint interactions C⋯H; g fingerprint interactions C⋯C

HS dnorm, interactions of non-classic H bonds, and fingerprints of compound BFQ. a Fingerprint contact C⋯H; b Ϙ2 interaction C18–H18⋯π with donor region (13) and receptor region (12); (c) fingerprint contact H⋯H; c, d Ϙ1 interaction C19-H19⋯O1 specifying the donor region (14) and the recipient region (15); e fingerprint contact O⋯ H; f fingerprint contact C⋯C
“Fingerprints” representations of contacts in which the compound is involved can be seen in Fig. 4 for the NFQ molecule and in Fig. 5 for the BFQ molecule. The HS analysis provides a unique environmental identification of each molecule throughout the combination of de and di contacts in a 2D graph, which indicates the contribution of each type of interaction observed in crystalline solid state [27, 43]. The distance from HS to the nearest outer atom (de) is displayed on the ordinate, while the distance from HS to the nearest inner atom (di) is displayed on the abscissa. For both structures, the highest percentage of interactions occurs between hydrogens, as can be seen in Figs. 4c and 5c, with values of 39.7% and 42.7% of H⋯H for NFQ and BFQ, respectively. This happens because organic compounds are almost always surrounded by hydrogens, and, therefore, the interactions between these atoms constitute a great percentage of the total interactions, as we can see at Ϙ1 NFQ and Ϙ2 BFQ interactions. On the other hand, non-classical hydrogen bonds from O⋯H interactions represent the fingerprint base with a total of 30.7% of the interactions for NFQ, represented by the Ϙ2 and Ϙ3 interaction, and only 17.1% for BFQ, represented by the Ϙ1 interaction. Figures 4b and 5e shows two sharp peaks (9), (10) and (16), (17), characteristic of O···H contacts. The upper peak (9), corresponding the atoms H15 and H20 in NFQ, and for (16) lies in regions where de > di corresponds to hydrogen donation sites in the H19 atom, while the lower peaks (10) and (17) are in regions referring to hydrogen acceptor sites [26, 27], i.e., the O3 and O5 atoms in NFQ and the O1 atom in BFQ. Therefore, for NFQ, the crystalline arrangement is preferentially stabilized by this type of arrangement.
Another useful piece of information given by fingerprints is about hydrophobic interactions present in the compound. When the C⋯H contacts are wings, as in Figs. 4f and 5a, this is a strong indication of the existence of C–H⋯π interactions. For BFQ, regions (12) and (13) correspond to a density of 18.7%, the most dominant contribution, which represents dimer formation and indicates that the arrangement is stabilized preferentially due to the C18–H18⋯π interaction. The upper peak (18) corresponds to hydrogen donation sites, i.e., H20 atom, and the lower peak (19) indicates the acceptor sites of hydrogen, i.e., ring B. However, this does not happen for NFQ, as is evident in Fig. 4e, where regions (3) and (4) and peaks (7) and (8), the C20–H20···π interaction, were clearly weaker than the others. Other information taken from fingerprints refers to the C⋯C interactions characterized by π⋯π type contacts. For both compounds, this type of interaction contributes the lowest percentage, 3% and 2.7%, respectively, seen in Figs. 4g and 5 f. The significantly low value found for π⋯π interactions demonstrates that this type of interaction is not the most important for packaging of the structures. This conclusion is evidenced by the lack of information regarding π⋯π contacts in the interactions seen by the software Mercury [24] and described in Table 2.
Molecular contacts can be, in addition, analyzed using a shape index map from HS. The map shows concave curvature as red regions representing the C–H⋯π interactions, while C–H donor regions are illustrated in blue and indicate the convex curvature with opposite sign. Besides that, π⋯π interactions are represented as “bowtie” shapes, indicating regions where two molecules meet. As can be seen, the stabilization of BFQ crystal packing occurs by Ϙ2 interaction C18–H18···Cg B [H···Cg 2.802 Å] illustrated in regions 21 and 22 of Fig. 5b. Also, NFQ is stabilized in crystal packing by the C20–H20···Cg B [H···Cg 2.851 Å] interaction demonstrated in Fig. 5a. The absence of a “bowtie” shape in both molecules indicates the structures that do not present π···π contact in the interactions, as we saw in the fingerprints and in Table 2 and Fig. 6.

HS shape index showing the C–H⋯π interactions established by the crystal packing of a NFQ and b BFQ
Previous studies have shown that 4(1H)-quinolinones have several uses as potential drugs. For example, the compound [6-bromo-1-(4-fluorophenylmethyl)-4(1H)-quinolinon-3-yl)]-4-hydroxy-2-oxo-3-butenoic acid (similar to NFQ and BFQ) acts as an HIV-1 integrase inhibitor. This occurs because the p-F-benzyl group points toward the catalytic loop responsible for the binding mode in the HIV-1 integrase enzyme [44]. Also, the similar compound 2-((ethyl(4-fluorobenzyl)amino)methyl)- 7,8-dimethylquinolin-4(1H)-one exhibits effects on Ca2+ mobilization in the 1321 N1 human astrocytoma cell line, and is considered as potential drug candidate for the treatment of cardiovascular disease [45]. Another example is the derivate of quinoline oxime sulfonic acids 7-chloro-2,3-dihydro-1-(2-methylbenzoyl)-4(IH)-quinolinone-4-oxime-o-sul fonic acid compound, which affects Na+ and K+ transport in distal nephron segments, including the cortical collecting duct and connecting tubule isolated from rabbit kidneys [46]. For the two quinolinones NFQ and BFQ, further analyses will be required to identify possible biological applications—analyses that are facilitated by structural deciphering. However, the nitro and bromine benzyl ligands in NFQ and BFQ molecules can act as good ring activator groups facilitating possible reactions in the structural body of quinolinone as an active agent in potential biological applications as demonstrated in previous studies.
Theoretical analysis
Table 3 presents IR intensities of the main groups for NFQ and BFQ, and their experimental and calculated vibrational frequencies (vibrational assignments). Furthermore, Fig. 7 presents the experimental and theoretical spectra (overlapped), and these results show satisfactory agreement between spectra, although shifts in energy are apparent in some cases. It should be noted that calculations were made for a free molecule in vacuum, while experiments were performed for solid samples. Furthermore, the anharmonicity is neglected in the real system for calculated vibrations [47]. This may explain disagreements between calculated and observed vibrational wave numbers.

Overloaded experimental (KBr) and theoretical infrared spectra of a NFQ and b BFQ
To avoid a long discussion of the many vibrational modes of these compounds, we have chosen to address the main constituent groups of each structure. The vibrations that occur at frequencies between 3050 cm−1 and 3010 cm−1 represent stretching of the CH bond [48]; results for this mode calculated in NFQ molecule are in the range of 2998 cm−1 to 3059 cm−1. Experimental values were in the range of 3091 cm−1 to 3057 cm−1, while in BFQ these values are 3038 cm−1 to 2989 cm−1 and 3053 cm−1 to 3065 cm−1, respectively. ν(C-C)aromatic stretch absorptions are reported at a band in 1600 cm−1 and another in 1475 cm−1, while in NFQ they occur at 1573 cm−1 and in 1476 cm−1; the corresponding theoretical results occur at 1603 cm−1 and in 1555 cm−1. For BFQ they occur at 1574 cm−1 experimentally, and at 1581 cm−1 theoretically. Vinyl group stretches occurs at 1630 ± 30 cm−1 or at lower frequencies in the case of conjugation with higher intensity [48]. In NFQ, the calculated value is 1598 cm−1, while in the experimental IR spectra it comes out at 1604 cm−1. For the BFQ molecule, this calculated stretch is 1601 cm−1 and the experimental stretch is at 1607 cm−1.
Conjugation of the carbonyl group with an aryl shifts the normal νC=O band to a lower frequency (1700–1675 cm−1) [48]. The calculated value for this mode is 1647 cm−1, while the experimental value is 1670 cm−1 for NFQ; on the other hand, BFQ presents these values at 1689 cm−1 (theoretical) and 1669 cm−1 (experimental). The ν(C–S) mode has variable intensity and could be found over a wide region [49]. Data reported in the literature also confirm this wide range of regions for compounds that have a benzene sulfonamide scaffold; in these cases the νC–S mode occurs at 944 cm−1 [50]; 833 cm−1 [51], 553 cm−1 [52], 666 cm−1 [53], etc. The calculated value for this band appears for NFQ at 696 cm−1, while the experimental value is 686 cm−1. The BFQ has theoretical vibration at 698 cm−1, in experimental spectra it is 685 cm−1.
The SO2 asymmetric and symmetric stretches occur at 1325 cm−1 and at 1140 cm−1 [48]. In DFT, these modes occur at 1276 cm−1 and 1042 cm−1. In experimental spectra of NFQ, these modes occur at 1309 cm−1 and 1143 cm−1. BFQ presents these values at 1293 cm−1 and 1048 cm−1 for theoretical calculations, and, in experimental spectra, at 1359 cm−1 and 1050 cm−1. The ν(S–N) vibration occurs at 905 ± 30 cm−1 [54]. Here, this mode occurs at 893 cm−1 (theoretical) and 898 cm−1 in experimental spectra for NFQ, while in BFQ these values are 908 cm−1 and 918 cm−1. The deformations of the NO2 group appear at low frequency [53], and, in the present study, only the NFQ molecule presents this group in its structure. The NO2 stretching vibrations, when linked at an aromatic ring, occur at 1520 ± 30 cm−1 (asymmetric) and 1335 ± 20 cm−1 (symmetric) [55]. Here, 1529 cm−1 and 1354 cm−1 are the values in IR spectra, respectively, while the calculated values are 1547 cm−1 and 1350 cm−1. The NO2 scissors occur at 850 ± 60 cm−1 when conjugated with aromatic molecules [48]. Here, 823 cm−1 (theoretical) and 831 cm−1 (experimental) bands are associated with the scissors mode.
Vibrations of C–halogen occur in the range of 1129–480 cm−1 [56, 57]. In aryl bromides, this absorption occurs between 1075 cm−1 and 1030 cm−1. The IR spectra of BFQ exhibit a band for this mode at 1012 cm−1, while the DFT value is 1113 cm−1. The molecular electrostatic potential (MEP) map, representing the charge distribution in a molecule, can be determined by computational methods [58]. Usually, red color indicates a negative region, which means it is susceptible to electrophilic attack; on the other hand, blue color indicates a positive region, where it is favorable for nucleophilic attack [59, 60]. The representation of calculated frontier molecular orbitals (HOMO and LUMO) and MEP for NFQ and BFQ are presented in Fig. 8.

Frontier molecular orbitals and molecular electrostatic potential (MEP) map of a NFQ and b BFQ
For NFQ, HOMO has a π*-bonding shape, and it appears alongside aromatic rings of the quinolinone backbone, while LUMO has a π-antibonding shape alongside carbonyl, vinyl, and the nitrobenzene ring. However, for compound BFQ, HOMO presents a π*-bonding shape, alongside aromatic rings of quinolinone backbone and bromobenzene, and LUMO has a π-antibonding shape, alongside carbonyl, vinyl and aromatic rings of the quinolinone backbone. For both compounds, the gap energy (EGap), hardness (η) and softness (σ) were calculated, according to the equations below [61]:
These values are 6.72 eV, 3.36 eV and 0.30 eV, respectively, for NFQ, while in BFQ they are 6.30 eV, 3.15 eV and 0.32 eV. All these values are summarized in Table 4 and, concerning Egap, they indicate that the existence of a strong deactivated group, NO2, in NFQ makes this compound slightly more kinetically stable and slightly less chemically reactive than BFQ, which presents a weaker deactivated group, Cl.
The hardness for NFQ is 3.36 eV, for BFQ is 3.15 eV, while the softness for NFQ is 0.30 eV and for BFQ is 0.32 eV. Since, in general terms, we can define hardness as a tendency for a charge transfer not to occur, while softness is a tendency to receive electrons [62], we can infer that NFQ has a higher resistance to changing its electronic configuration, while BFQ has a higher capacity to receive electrons. In addition, the electronic properties for the parent quinolone molecule (4DH) were calculated. So, the geometric parameters obtained from crystallographic data [63] were used as input to the quantum calculations and may help in the understanding of the substituent effect on the different properties. According to the results (Table 4) we may infer that the quinolone backbone has strong domain in the electronic properties, so that the effect of the substituents attached to it do not cause major changes. In MEP, the areas of high electron density (red in Fig. 8) are in sites with O atoms (SO2, C=O and NO2 groups). These are therefore electrophilic sites, while the areas of high depleted electron density (blue in Fig. 8) are in aromatic ring regions.
Conclusions
The hybrid compounds NFQ and BFQ are 1–4 quinolinones derivatives having a common backbone with different substituents. The connection with C10 is a m-nitro group in NFQ, packing in an orthorhombic group, Pbca, and a p-bromine group in BFQ pack into a monoclinic group, P21/n. On binding angles, it is notable that the molecular architecture presents a tetrahedral geometry, and a trigonal plane in different parts of the molecules. Also, no strong intramolecular interactions were found, with only weak intermolecular interactions occurring, which stabilize the crystalline packing: three interactions for NFQ and two for BFQ. The most important interaction contributing to packing stabilization in the BFQ molecule is C18–H18⋯π, and in NFQ it is C15–H15⋯O3. The experimental and theoretical spectra show satisfactory agreement between spectra. For NFQ, the HOMO has a π*-bonding shape along the aromatic rings of the quinolinone backbone and LUMO has a π-antibonding shape along the carbonyl, vinyl and nitrobenzene ring, while, for compound BFQ, HOMO presents π*-bonding shape, along aromatic rings of quinolinone backbone and bromobenzene, and LUMO has π-antibonding shape, along of carbonyl, vinyl and aromatic rings of quinolinone backbone.
References
Rodrigues-Silva C, Maniero MG, Peres MS, Guimarães JR (2014) Ocorrência e degradação de quinolonas por processos oxidativos avançados. Quim Nova 37:868–885
Lesher GY, Froelich EJ, Gruett MD, Bailey JH, Brundage RP (1962) 1,8-Naphthyridine derivatives. A new class of chemotherapeutic agents. J Med Chem 5(5):1063–1065
Andriole VT (2005) The quinolones: past present, and future. Clin Infect Dis 41:S113–S119
Shiro T, Fukaya T, Tobe M (2015) The chemistry and biological activity of heterocycle-fused quinolinone derivatives: a review. Eur J Med Chem 97:397–408
De Leon P, et al (2009) Quinolinone PDE2 Inhibitors. World Patent application WO2011011312A1
Saji H, Kimura H, Ono M, Matsumoto H (2014) Radioactive quinolinone derivative and pharmaceutical drug comprising the same. Japanese Patent CA2836400A1, 27 June 2014
Fatheree PR, Turner SD, Goldblum AA, Chao RS, Genov D (2016) Crystalline form of a quinolinone-carboxamide compound. US Patent US14816237
Cai S et al (2012) Methods for synthesizing quinolinone compounds. US Patent application US20050137399A1
Roussaki M et al (2013) Synthesis and anti-parasitic activity of a novel quinolinone-chalcone series. Bioorg Med Chem Lett 23:6436–6441
Kwak SH et al (2015) Solid-phase synthesis of quinolinone library. ACS Comb Sci 17:60–69
Kwak SH et al (2016) Discovery and structure–activity relationship studies of quinolinone derivatives as potent IL-2 suppressive agents. Bioorg Med Chem 24:5357–5367
Lakshmi Narayana Sharma K et al (2017) Palladium-catalyzed domino sequence for the synthesis of N-aryl quinolinone-3-carboxylate derivatives and their anti-proliferative activity. Tetrahedron Lett 58:1127–1131
Nanke Y et al. (2016) Rebamipide, an amino acid analog of 2(1H)-quinolinone, inhibits the formation of human osteoclasts. Biomed Res Int 2016:6824719
Kalkhambkar RG et al (2012) ChemInform Abstract: Synthesis and biological studies of some new acrylic acid ethyl esters of quinolinone. Monatshefte für Chemie − Chemical Monthly 143:1075–1086
Vats P et al (2014) Chromenone and quinolinone derivatives as potent antioxidant agents. Med Chem Res 23:4907–4914
Rylova G et al (2012) Abstract 4748: molecular targets of quinolinone derivatives with anticancer activity. Cancer Res 72:4748 LP–4744748
Rylova G et al (2014) Abstract 4624: molecular target identification of quinolinone based anticancer compounds. Cancer Res 74:4624 LP–4624624
Paramaguru G, Vijay Solomon R, Jagadeeswari S, Venuvanalingam P, Renganathan R (2014) Tuning the photophysical properties of 2-quinolinone-based donor-acceptor molecules through N-versus O-alkylation: insights from experimental and theoretical investigations. Eur J Org Chem 2014:753–766
Wiles JA, Bradbury BJ, Pucci MJ (2010) New quinolone antibiotics: a survey of the literature from 2005 to 2010. Expert Opin Ther Pat 20:1295–1319
Desiraju GR (2014) Crystallography and geopolitics. Science 343:1057–1057
Bruker-AXS (2014) APEX2. https://www.bruker.com/products/x-ray-diffraction-and-elemental-analysis/single-crystal-x-ray-diffraction/sc-xrd-software/overview/sc-xrd-software/apex3.html
Sheldrick GM (2015) Crystal structure refinement with SHELXL. Acta Cryst 71:3–8
Farrugia LJ (2012) WinGX and ORTEP for windows: an update. J Appl Crystallogr 45:849–854
Macrae CF et al (2006) Mercury: visualization and analysis of crystal structures. J Appl Crystallogr 39:453–457
Spek AL (2003) Single-crystal structure validation with the program PLATON. J Appl Crystallogr 36:7–13
McKinnon JJ, Spackman MA, Mitchell AS (2004) Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Cryst 60:627–668
Spackman MA, McKinnon JJ (2002) Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 4:378–392
Spackman MA, Jayatilaka D (2009) Hirshfeld surface analysis. CrystEngComm 11:19
Wood PA, McKinnon JJ, Parsons S, Pidcock E, Spackman MA (2008) Analysis of the compression of molecular crystal structures using Hirshfeld surfaces. CrystEngComm 10:368–376. https://doi.org/10.1039/b715494a
Chattopadhyay B et al (2010) Supramolecular architectures in 5,5′-substituted Hydantoins: crystal structures and Hirshfeld surface analyses. Cryst Growth Des 10:4476–4484
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Accounts 120:215–241
Paciotti R et al (2015) Serine O-sulfation probed by IRMPD spectroscopy. Phys Chem Chem Phys 17:25891–25904
Foresman JB, Frisch AE (1996) Exploring chemistry with electronic structure methods. Gaussian, Inc., Wallingford, CT
Buczek A, Kupka T, Broda MA, Żyła A (2016) Predicting the structure and vibrational frequencies of ethylene using harmonic and anharmonic approaches at the Kohn–sham complete basis set limit. J Mol Model 22:42
Jamroz MH (2010) Vibrational energy distribution analysis. VEDA 4, Warsaw. http://smmg.pl/index.php/software/sowtware-veda.html.
Dennington R, Keith T, Millam J (2009) GaussView, ver 5. Semichem Inc., Shawnee Mission, KS.
Silva NC (2013) Caracterização esrutural do flavonoide C16H14O3. Masters thesis, Universidade Estadual de Goiás, Brazil
Kupcewicz B et al (2013) Structure-cytotoxic activity relationship of 3-arylideneflavanone and chromanone (E,Z isomers) and 3-arylflavones. Bioorg Med Chem Lett 23:4102–4106
Zimmerman JR et al (2015) The synthesis of a new class of highly fluorescent chromones via an inverse-demand hetero-Diels-Alder reaction. Org Lett 17:3256–3259
Dziewulska-Kułaczkowska A, Mazur L (2011) Structural studies and characterization of 3-formylchromone and products of its reactions with chosen primary aromatic amines. J Mol Struct 985:233–242
Krupa J, Lackner H, Jones PG, et al (1989) The Absolute Configuration of the Juglomycins. Z Naturforsch B 44:345–352
Brandl M, Weiss MS, Jabs A, Suhnel J, Hilgenfled R (2001) C–H⋯pi-interactions in proteins. J Mol Biol 307:357–377
McKinnon JJ, Jayatilaka D, Spackman MA (2007) Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem Commun 0:3814–3816. doi:https://doi.org/10.1039/b704980c
Vandurm P et al (2009) Structural and theoretical studies of [6-bromo-1-(4-fluorophenylmethyl)-4(1H)-quinolinon-3-yl]-4-hydroxy-2-oxo-3-butenoïc acid as HIV-1 integrase inhibitor. Bioorg Med Chem Lett 19:4806–4809
Sakuma K, Nakagawa H, Oikawa T, Noda M, Ikeda S (2017) Effects of 4(1H)-quinolinone derivative, a novel non-nucleotide allosteric purinergic P2Y2agonist, on cardiomyocytes in neonatal rats. Sci Rep 7:1–10
Yasoshima K, Yamasaki F, Shinkawa T, Yoshitomi K, Imai M (1993) Effect of a novel diuretic, 7-chloro-2,3-dihydro-1-(2-methylbenzoyl)-4(IH)-quinolinone-4-oxime-o- sulfonic acid, potassium salt (M17055) on Na+ and K+ transport in the distal nephron segments. J Pharmacol Exp Ther 266:1581 LP–1581588
Karabacak M, Çınar M, Çoruh A, Kurt M (2009) Theoretical investigation on the molecular structure, infrared, Raman and NMR spectra of Para-halogen benzenesulfonamides, 4-X-C6H4SO2NH2 (X=Cl, Br or F). J Mol Struct 919:26–33
Pavia DL, Lampman GM, Kriz GS, Vyvyan JA (2015) Introduction to spectroscopy, 4th edn. Cengage Learning, Boston, MA
Benhalima N et al (2016) Solvent effects on molecular structure, vibrational frequencies, and NLO properties of N-(2,3-dichlorophenyl)-2-nitrobenzene–sulfonamide: a density functional theory study. Braz J Phys 46:371–383
Vinod KS, Periandy S, Govindarajan M (2016) Spectroscopic [FT-IR and FT-Raman] and molecular modeling (MM) study of benzene sulfonamide molecule using quantum chemical calculations. J Mol Struct 1116:226–235
Chohan ZH, Youssoufi MH, Jarrahpour A, Ben Hadda T (2010) Identification of antibacterial and antifungal pharmacophore sites for potent bacteria and fungi inhibition: indolenyl sulfonamide derivatives. Eur J Med Chem 45:1189–1199
Chandran A et al (2012) FT-IR, FT-Raman and computational study of (E)-N-carbamimidoyl-4-((4-methoxybenzylidene)amino)benzenesulfonamide. Spectrochim Acta A Mol Biomol Spectrosc 92:84–90
Sarojini K, Krishnan H, Kanakam CC, Muthu SS (2013) Structural, spectroscopic studies, NBO analysis, NLO and HOMO–LUMO of 4-methyl-N-(3-nitrophenyl)benzene sulfonamide with experimental and theoretical approaches. Spectrochim Acta A Mol Biomol Spectrosc 108:159–170
Roeges NPG (1994) A guide to the complete interpretation of infrared spectra of organic structures. Wiley, New York
Vaz WF et al (2016) Synthesis, characterization, and third-order nonlinear optical properties of a new neolignane analogue. RSC Adv 6:79215–79227
Mooney EF (1963) The infrared spectra of chloro- and bromobenzene derivatives—I. Anisoles and phenetoles. Spectrochim Acta 19:877–887
Mooney EF (1964) The infra-red spectra of chloro- and bromobenzene derivatives—II. Nitrobenzenes. Spectrochim Acta 20:1021–1032
Naray-Szabo G, Ferenczy GG (1995) Molecular electrostatics. Chem Rev 95:829–847
Galabov B, Nikolova V, Ilieva S (2013) Does the molecular electrostatic potential reflect the effects of substituents in aromatic systems? Chem Eur J 19:5149–5155
Rahmani R, Boukabcha N, Chouaih A, Hamzaoui F, Goumri-Said S (2018) On the molecular structure, vibrational spectra, HOMO-LUMO, molecular electrostatic potential, UV–Vis, first order hyperpolarizability, and thermodynamic investigations of 3-(4-chlorophenyl)-1-(1yridine-3-yl) prop-2-en-1-one by quantum chemistry calculations. J Mol Struct 1155:484–495
Oliveira SS et al (2017) Synthesis, characterization, and computational study of the supramolecular arrangement of a novel cinnamic acid derivative. J Mol Model 23:35
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105:7512–7516
Nasiri HR, Bolte M, Schwalbe H (2006) Tautomerism of 4-hydroxy-4(1H)quinolone. Heterocycl Commun 12:319–322
Acknowledgments
The authors would like to thank Brazilian funding agencies Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Fundação de Amparo à Pesquisa do Estado de Goiás (FAPEG) for financial support and fellowships. Research developed with support of the High-Performance Computing Center at the Universidade Estadual de Goiás (UEG). Single crystal X-ray diffraction data were collected at Universidade de São Paulo (USP).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Michelini, L.J., Vaz, W.F., D’Oliveira, G.D.C. et al. Analysis of two novel 1–4 quinolinone structures with bromine and nitrobenzyl ligands. J Mol Model 25, 55 (2019). https://doi.org/10.1007/s00894-019-3937-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-3937-3