
Abstract
In this study, we propose new armchair single-walled nanotubes (SWNTs) for stable adsorption, increasing drug delivery performance and decreasing side effects of pro-carbazine (Pro-CB) anti-cancer in the framework of B3LYP/6-31 g*/Lanl2DZ level of theory. Indeed, doping gallium (Ga) metal in SWNTs is naturally followed by changing of geometry, increasing dipole moment, and creating one site with high reactivity in order to better adsorption of the drug molecule. Chemical reactivity descriptors show that SWNTs and Pro-CB have electrophile and nucleophile roles in interaction, respectively. More importantly, high local and dual softness in Ga-doped SWNTs indicate improvement of drug adsorption. Parallel and perpendicular complexes result from their interaction in the N and the O sites. Negative values of binding energy (Ebind) show that composed complexes are energetically stable especially in the O site in comparison with the N site. On the other hand, more negative value of the Ebind in SWCNTs shows that these nanotubes are more effective for drug adsorption than their boron nitride counterparts.

The Ga dopping results in reducing of HOMO-LUMO gap and increasing charge transfer between SWNTs and Pro-CB, and formation better complex, especially SWCNT.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Single-walled nanotubes (SWNTs) are considered a novel class of carriers with significant efficiency in drug-delivery systems [1]. Thanks to their structural properties, such as high surface area, specific size, highly site-selective delivery, and sensitivity [2,3,4,5,6,7], SWNTs have the ability to transport drug molecules to target cells without side effects [4, 6,7,8]. Indeed, this release of drug molecules is due to better adsorption of drug ligands to the in/out side of SWNTs. Nowadays, carbon and boron nitride nanotubes (CNTs and BNNTs) are strong candidates in medical and biological fields [9,10,11,12,13,14,15,16,17]. Electrostatic potentials of these nanotubes in outer and inner surfaces cause their different behavior in interaction with other molecules [18, 19]. This is why we can see the trace of many research groups in a vast variety of theoretical and experimental studies [2, 3, 20,21,22,23,24,25,26,27].
The in vitro and in vivo studies showed that using SWCNTs causes selective transport of cisplatin (cis-DDP) to the tumor cells and reducing side effects [1, 28, 29]. In other words, high efficiency of interaction of SWCNTs and platinum complexes anti-cancer was showed that this nanotubes have not toxicity [21, 30]. The interaction between SWCNTs and folic acid has been studied experimentally [31]. Using SWCNT carriers results in decreasing interaction of folic acid with other molecules and safe delivery by folate receptors [20]. In another work, adsorption of leflunomide on SWCNTs and SWBNNTs has been determined in order to introduce these nanotubes as multipurpose innovative carriers for better drug delivery and diagnostic application [27].
It has been observed that doping of metal on SWNTs results in better adsorption and reduction of possible toxicity such as the one in platinum-based drugs on Al-doped BNNTs [32, 33]. Sensitivity of gold doped on CNT (AuNPs) to environmental factors such as pH has been investigated [34]. Existence of intermolecular forces between SWNTs and drug molecules conveniently are assigned using quantum chemical calculations.
The above-reported studies aimed at introducing SWNTs as good deliverers of various drug molecules. However, the reactivity and adsorption of pro-carbazine (Pro-CB) drug on SWNTs carrier have not attracted much attention yet. In this study, we are going to investigate theoretical adsorption of Pro-CB drug as brain anti-cancer on armchair (5, 5) SWXNTs (X = C, BN)]. Geometries of these structures with a single bond are presented in Fig. 1. In order to improve Pro-CB adsorption and increasing chemical reactivity in these nanotubes, one gallium atom was doped in SWNTs. Therefore, our main objective is determination of the ground-state structures, chemical reactivity, and origin of these linkages and nature of adsorptions using density functional theory (DFT) method. In the following, natural bond orbital (NBO) [35] analysis was carried out to identify the underlying nature of events in their interactions.

Optimal structures of carbon [C] (a), carbon/gallium [C/Ga] (b), pro-carbazine [Pro-CB] (c), boron nitride [BN] (d), and boron nitride/gallium [BN/Ga] (f) armchair (5, 5) nanotubes, respectively
Computational methods
Geometry optimizations, calculations of chemical reactivity descriptors, electronic analysis of SWXNTs (X = C, BN) and Pro-CB compounds were carried out by using Gaussian 09 program package [36] in the framework of density functional theory (DFT) and 6–31 + g(d)/Lanl2DZ basis set for light/heavy atoms [37,38,39]. To confirm that the structures refer to the corresponding local minima, vibrational frequencies were calculated and no imaginary frequency was obtained for any structures such as in other reports [40]. Further, the stability of the systems was evaluated through computing energies of binding (Ebind):
In these equations, Ecomplex, ESWNT, and Edrug are the total energy of the complex, SWNTs, and Pro-CB anti-cancer. Tendency of an electron in a compound to interact with other molecules is called chemical reactivity [41]. In the framework of DFT method, global and local indices of reactivity are calculated using ionization potential (IP) and electron affinity (EA) [42]:
Global descriptors are electronic chemical potential (μ), chemical hardness (η), chemical softness (S), and electrophilicity (ω), respectively. Escaping tendency of electrons from the equilibrium systems is called chemical potential [41]:
Resistance of a system to electron transfer is hardness; its large value shows that the system is harder and more stable or less reactive [41, 43]:
Reverse of hardness is global softness and its large value indicates less stability and high tendency to reacts with other molecules [41]:
Exchange of electrons is local electrophilicity (ω) [44]:
Favorable site of SWNTs in interaction with Pro-CB anti-cancer are predicted by investigation of local descriptors such as Fukui function (f), softness (s), philicity (ω), dual softness (∆s), and dual philicity (∆ω). Sensitivity of the chemical potential of a system to a local external potential is called Fukui function (f); it is used in order to characterize reactivity of an atom in a compound [41]. Finite difference approximation has been applied to determine nucleophilic (f+) and electrophilic (f−) attacks [41]:
where \( {q}_k^N \), \( {q}_k^{N+1} \) , and \( {q}_k^{N-1} \) are electronic populations of the N, (N + 1) and (N-1) electron systems on selected atom, respectively. Role a site in a compound in electrophilic (nucleophilic) attacks identify by using local softness and philicity [45]:
when α = + , − means nucleophilic, electrophilic attacks; maximum positive value of local softness in the k site indicate that this site has most electrophile property in compound and it is favorable for nucleophilic attack in reaction with Pro-CB. Selectivity of the k site in nucleophilic (electrophilic) attacks is evaluated by dual local softness [46, 47]:
the ∆sk > 0 and ∆sk < 0 mean site k is favorable for nucleophilic and electrophilic attacks, respectively.
Moreover, geometrical information is completed by the results of NBO analysis [35]. Type of bonds, depletion of occupancies, percent of Lewis and non-Lewis, and stabilization energies [35, 48] are obtained by NBO analysis. The stabilization energy (E2ij) in Eq. (12) includes interaction Hamiltonian \( \widehat{\mathrm{H}} \), orbital energies Ej and Ei and also \( \left\langle \mathrm{i}\vert \widehat{\mathrm{H}}\vert \mathrm{j}\right\rangle \) that is the matrix element.
Results and discussion
Geometric and energetic results
Starting point theoretical studies is determination most stable geometries. Therefore, armchair (5, 5) single-walled nanotubes (SWXNT, X = C and BN) with finite length and hydrogen saturation in both ends were optimized without any symmetry constrains to obtain local minimum of structures (see Fig. 1). There is no imaginary frequency in any of the SWNTs structures, so they are in real minima.
Doping of gallium metal in SWNTs is naturally followed by a change of carbon and boron NT geometries; for example, C─Ga and N─Ga bonds length average 2.01 and 1.84 Å, respectively. Elongation of bonds rather than C─C (1.44 Å) and N─B (1.45 Å) [49, 50] partly result in an increase of dipole moment and creation of a favorable active site in order to better adsorption and more effective interaction with Pro-CB drug. Therefore, chemical reactivity descriptors can set the stage to understand the behavior of SWNTs in their interactions.
Global reactivity descriptors can be used in order to describe stability and role of molecules in reaction (see Table 1. According to global reactivity indexes, more negative values of the μ in SWNTs structures rather than Pro-CB showing easily electron acceptance and playing an electrophile role in interactions; so, Pro-CB acts as a nucleophile. On the other hand, the η and S decreases and increases by doping of gallium in SWNTs, so doped-SWNTs have less stability and more reactivity than pure SWNTs. In addition, Ga-SWNTs have better ability for acceptation of electron due to increasing the ω in comparison with SWNTs.
Awareness of active sites in SWNTs and Pro-CB helps to better understand interactions. Therefore, we have investigated active sites in nucleo (electro) phile using local softness (s+and s−). Our results show that more positive value of s+ and ∆s in the Ga doped SWNTs centers, especially Ga atom cause Pro-CB attacks with positive value of the s− to these centers from the N and O sites, see Table 2. According to this table, nucleophile attacks of Pro-CB to Ga-SWNTs carried out from the O site due to high local softness and dual softness in this center.
One of the other significant factors to show stability of SWNTs is highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) gap. Increasing HOMO–LUMO gap with gallium doping partly results in decreasing of HOMOPro-CB-LUMOSWNTs gaps (see Table 1 and Fig. 2). Consequently, charge transfer, interaction, and delivery of Pro-CB anti-cancer drug probably carried out better than pure SWNTs.

HOMO-LUMO gap of SWXNTs (X = C, BN) compounds are obtained using B3LYP/6-31 g*/Lanl2DZ level of theory
In the following, SWNTs has interacted with Pro-CB molecules at parallel and perpendicular orientation from the nitrogen (N) and oxygen (O) sites, respectively, as shown in Fig. 3. Optimized structural parameters of SWNTs-Pro-CB complexes are given in Table 3. The O─X (X = C, B, and Ga) distances in perpendicular configurations are lower than N─X (X = C, B, and Ga) the planar ones, which reflected by the corresponding higher binding energies. Low bond length results in better adsorption of drug on SWNTs, low side effects, and better drug delivery.

Low-lying structures of SWNT/Pro-carbazine complexes with dipole moment (μ, Debye), dihedral angle (°, degrees), distances of nitrogen (N) and oxygen (O) sites with SWNTs (d, Å), respectively
The X─O (N)─Ph bond angles in the O site are larger than the N site complexes; this issue indicates that perpendicular complexes are more stable than ones. Besides, the X─O (N)─C─C dihedral angles in the O site are larger than the N site, which can be due to repulsive interaction between N─H bond of Pro-CB and surface of SWNTs in parallel orientation. Therefore, binding of Pro-CB to SWNTs in the perpendicular complex is more favorable owing to low distances, bond angles, repulsive interaction, and dihedral angles.
Density of state (DOS)
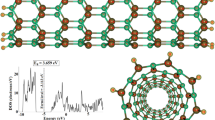
One of the reliable methods to analyze the chemical structures from an electronic viewpoint is density of state (DOS). Adsorption of Pro-CB anti-cancer on SWNTs leads to small modification of drug. We have evaluated the DOS for the best orientation of adsorption in order to confirm preferential adsorption and charge transfers (see Fig. 4 and Fig. S1) (S means Supplementary file). The DOS plots show some discrepancies in the height and slight shifts of energy level of SWNTs; this issue indicates that the drug molecule adsorbs on SWNTs. According to Fig. 4, more discrepancy of occupied and virtual orbitals (≈5.0 eV) in the BNNTs than CNTs (≈3.0 eV) mainly cause low charge transfers and weak chemical adsorption and defective drug delivery than ones.

Calculated electronic density of states (DOS) for the SWXNTs (X = C, BN) and pro-carbazine (Pro-CB) drug as a function of the energy
NBO analysis
Structural and energetical information is completed using natural bond orbital (NBO) theory [35]. Decreasing negative charges of the N and O atoms in both planar and perpendicular complexes show that charge transfer is carried out from Pro-CB to SWNTs (see Table 4). A decrease in the positive charge value of the Ga metal is owing to the delocalization of the N and O lone pairs to the NBO acceptors. Therefore, increasing of electronic charge of metal in the Ga-SWXNTs (X = C, BN) showing drug stabilization on their surfaces in order to better drug delivery. Charge transfer values in perpendicular complexes from O site are more than the N site of parallel ones. There is a relation between the charge transfer and binding energies, shown in Fig. 5. For example, the {Pro-CB[Ga-SWXNT (X = C, BN)]} compounds in the O site with high Ebind (− 138.2, − 45.6 kcal mol−1) have maximum charge transfer (69.6, 70.9 kcal mol−1), respectively.

Correlation of charge transfer (CT) and binding energy (Ebind) in the N and O sites of adsorption in the complexes
There are two types of weak donation/back-donation interactions in pure SWNT complexes, which consist of Pro-CB → SWNT and SWNT → Pro-CB (as shown in Table 5). Therefore, these donor–acceptor interactions cause weak intermolecular interactions between the Pro-CB anti-cancer and pure nanotubes and very low Ebind in this adsorption. Also, NBO results show that the Ga-SWNT-[Pro-CB] complexes have significant donor–acceptor interactions with higher stabilization energies in the O site than in the N site (see Table 5). Therefore, interaction in the O site is stronger than in the N site, which is in good agreement with reducing occupancy, Ebind, and DOS analysis.
Conclusions
In this study, SWXNTs (X = C, BN) and Pro-CB structures were optimized and the most favorable orientation in their interactions were investigated. Our results show that doping of gallium atom decreases HOMOPro-CB and LUMOSWXNT gap and increases donor–acceptor interactions. These issues indicate better adsorption of Pro-CB on these doped nanotubes, especially Ga-SWCNT. In addition, its Ebind value is more negative than SWCNT. Therefore, among SWNTs, Ga-doped SWCNTs is considered as a suitable carrier to transport drug molecules from the O site.
References
Flahaut E (2011) Carbon nanotubes for biomedical applications. Springer, Berlin
Lu X, Tian F, Xu X, Wang N, Zhang Q (2003) A theoretical exploration of the 1,3-dipolar cycloadditions onto the sidewalls of (n,n) armchair single-wall carbon nanotubes. J Am Chem Soc 125:10459–10464
Peles-Lemli B, Kelterer A, Fabian W, Kunsági-Máté S (2010) Noncovalent interaction between aniline and carbon nanotubes: effect of nanotube diameter and the hydrogen-bonded solvent methanol on the adsorption energy and the photophysics. J Phys Chem C 114:5898–5905
Kam NWS, Dai H (2005) Carbon nanotubes as intracellular protein transporters: generality and biological functionality. J Am Chem Soc 127:6021–6026
Saito R, G. Dresselhaus, M. S. Dresselhaus (1998) Physical properties of carbon nanotubes. World Scientific. https://doi.org/10.1142/p080
Liu Z, Chen K, Davis C, Sherlock S, Cao Q, Chen X, Dai H (2008) Drug delivery with carbon nanotubes for in vivo cancer treatment. Cancer Res 68:6652–6660
Liu Z, Winters M, Holodniy M, Dai H (2007) siRNA delivery into human T cells and primary cells with carbon nanotube transporters. Angew Chem Int Ed 46:2023–2027
Bhirde A, Gavard J, Zhang G, Sousa A, Masedunskas A (2009) Targeted killing of cancer cells in vivo and in vitro with EGF-directed carbon nanotube-based drug delivery. ACS Nano 3:307–316
Adeli M, Soleyman R, Beiranvand Z, Madani F (2013) Carbon nanotubes in cancer therapy: a more precise look at the role of carbon nanotube–polymer interactions. Chem Soc Rev 42:5231–5256
Elsaesser A, Howard CV (2012) Toxicology of nanoparticles. Adv Drug Deliv Rev 64:129–137
Prakash S, Malhotra M, Shao W, Tomaro-Duchesneau C, Abbasi S (2011) Polymeric nanohybrids and functionalized carbon nanotubes as drug delivery carriers for cancer therapy. Adv Drug Deliv Rev 63:1340–1351
Iijima S (1991) Helical microtubules of graphitic carbon. Nature 354:56–58
Vashist SK, Zheng D, Pastorin G, Al-Rubeaan K, Luong JHT, Sheu F-S (2011) Delivery of drugs and biomolecules using carbon nanotubes. Carbon 49:4077–4097
Chen X, Wu P, Rousseas M, Okawa D, Gartner Z, Zettl A (2009) Boron nitride nanotubes are noncytotoxic and can be functionalized for interaction with proteins and cells. J Am Chem Soc 131:890–891
Rouse JG, Yang J, Barron AR, Monteiro- Riviere N (2006) A fullerene-based amino acid nanoparticle interactions with human epidermal keratinocytes. Toxicol in Vitro 20:1313–1320
Monteiro-Riviere NA, Nemanich RJ, Inman AO, Wang YY, Riviere JE (2005) Multiwalled carbon nanotube interactions with human epidermal keratinocytes. Toxicol Lett 155:377–384
Shvedova A, Castranova V, Kisin E, Schwegler-Berry D, Murray A, Gandelsman V (2003) Exposure to carbon nanotube material: assessment of nanotube cytotoxicity using human keratinocyte cells. J Toxicol Environ Health A 66:1909–1926
Peralta-Inga Z, Lane P, Murray JS, Boyd S, Grice ME, O’Connor CJ, Politzer P (2003) E characterization of surface electrostatic potentials of some (5,5) and (n,1) carbon and boron/nitrogen model nanotubes. Nano 3:21–28
Politzer P, Lane P, Murray JS, Concha MC (2003) Comparative analysis of surface electrostatic potentials of carbon, boron/nitrogen and carbon/boron/nitrogen model nanotubes. J Mol Model 11:1–7
Castillo J, Svendsen WE, Rozlosnik N, Escobar P, Martínez F, Castillo-Leon J (2013) Detection of cancer cells using a peptide nanotube–folic acid modified graphene electrode. Analyst 138:1026–1031
Dhar S, Liu Z, Thomale J, Dai H, Lippard SJ (2008) Targeted single-wall carbon nanotube-mediated Pt(IV) prodrug delivery using folate as a homing device. J Am Chem Soc 130:11467–11476
Mavrandonakis A, Farantos SC, Froudakis GE (2006) Glycine interaction with carbon nanotubes: an ab initio study. J Phys Chem B 110:6048–6050
Zanella I, Fagan SB, Mota R, Fazzio A (2007) Ab initio study of pristine and Si-doped capped carbon nanotubes interacting with nimesulide molecules. Chem Phys Lett 439:348–353
Liu H, Bu Y, Mi Y, Wang Y (2009) Interaction site preference between carbon nanotube and nifedipine: a combined density functional theory and classical molecular dynamics study. J Mol Struct 901:163–168
de Leon A, Jalbout AF, Basiuk VA (2008) SWNT–amino acid interactions: a theoretical study. Chem Phys Lett 457:185–190
Hafizi H, Najafi Chermahini A, Mohammadnezhad G, Teimouri A (2015) A theoretical study on the interaction of amphetamine and single-walled carbon nanotubes. Appl Surf Sci 329:87–93
Raissi H, Mollania F (2014) Immunosuppressive agent leflunomide: a SWNTs-immobilized dihydroortate dehydrogenase inhibitory effect and computational study of its adsorption properties on zigzag single-walled (6,0) carbon and boron nitride nanotubes as controlled drug delivery devices. Eur J Pharm Sci 56:37–54
Ajima K, Yudasaka M, Murakami T, Maigne A, Shiba K, Ijima S (2005) Carbon nanohorns as anticancer drug carriers. Mol Pharm 2:475–480
Guven A, Rusakova IA, Lewis MT, Wilson LJ (2012) Cisplatin@US-tube carbon nanocapsules for enhanced chemotherapeutic delivery. Biomaterials 33:1455–1461
Dhar S, Daniel WL, Giljohann DA, Mirkin CA, Lippard SJ (2009) Polyvalent oligonucleotide gold nanoparticle conjugates as delivery vehicles for platinum(IV) warheads. J Am Chem Soc 131:14652–14653
Castillo JJ, Rozo CE, Castillo-Leon J, Rindzevicius T, Svendsen WE, Rozlosnik N, Anja Boisen N, Martinez F (2013) Computational and experimental studies of the interaction between single-walled carbon nanotubes and folic acid. Chem Phys Lett 564:60–64
Shakerzadeh E, Noorizadeh S (2014) A first principles study of pristine and Al-doped boron nitride nanotubes interacting with platinum-based anticancer drugs. Phys E 57:47–55
Karadas N, Ozakan SA (2014) Electrochemical preparation of sodium dodecylsulfate doped over-oxidized polypyrrole/multi-walled carbon nanotube composite on glassy carbon electrode and its application on sensitive and selective determination of anticancer drug: Pemetrexed. Talanta 119:248–254
Li J, Yoong SL, Goh WJ (2015) In vitro controlled release of cisplatin from gold-carbon nanobottles via cleavable linkages. Int J Nanomedicine 10:7425–7441
Foster J, Weinhold F (1980) Natural hybrid orbitals. J Am Chem Soc 102:7211–7218
M. J. Frisch (2009) In; Wallingford: Gaussian, Inc.
McLean A, Chandler G (1980) Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z= 11–18. J Chem Phys 72:5639–5648
Tirado-Rives J, Jorgensen WL (2008) Performance of B3LYP density functional methods for a large set of organic molecules. J Chem Theo Comput 4:297–306
Wadt WR, Hay PJ (1985) Ab initio effective core potentials for molecular calculations - potentials for main group elements Na to bi. J Chem Phys 85:284–298
Pakiari AH, Eshghi F (2017) Geometric and electronic structures of vanadium sub-nano clusters, Vn (n = 2-5), and their adsorption complexes with CO and O2 ligands: a DFT-NBO study. Phys Chem Res 5:601–615
Parr RG, Yang W (1989) Density-functional theory of atoms and molecules. Oxford University Press, New York Chapter 3 to 5
Mulliken RS (1934) A new electroaffinity scale; together with data on valence states and on valence ionization potentials and electron affinities. J Chem Phys 2:782–793
Parr RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105:7512–7516
Chattaraj PK, Sarkar U, Roy DR (2006) Update 1 of: Electrophilicity index. Chem Rev 106:2065
Lee C, Yang W, Parr RG (1988) Local softness and chemical reactivity in the molecules CO, SCN− H2CO. J Mol Struct 163:305–313
Parthasarathi R, Padmanabhan J, Elango M, Subramanian V, Chattaraj PK (2004) Intermolecular reactivity through the generalized philicity concept. Chem Phys Lett 394:225–230
Padmanabhan J, Parthasarathi R, Subramanian V, Chattaraj PK (2006) Chemical reactivity indices for the complete series of chlorinated benzenes: solvent effect. J Phys Chem A 110:2739–2745
Reed AE, Weinstock RB, Weinhold F (1998) Natural population analysis. J Chem Phys 83(1985):735–746
Gao G, Cagin T, Goddard WA (1998) Energetics, structure, mechanical and vibrational properties of single-walled carbon nanotubes. Nanotec 9:184
Terrones MJ, Romo-Herrera M, Cruz-Silva E, López-Urías F, Muñoz-Sandoval E, Velázquez-Salazar JJ, Terrones H, Bando Y, Golberg D (2007) Pure and doped bonitride nanotubes. Mater Today 10:30–38
Acknowledgements
In this work, Dr. Fazlolah Eshghi always has helpful hints, a lot of tips and suggestions for our work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 975 kb)
Rights and permissions
About this article

Cite this article
Ghoreishi, R., Kia, M. Chemical reactivity and adsorption properties of pro-carbazine anti-cancer drug on gallium-doped nanotubes: a quantum chemical study. J Mol Model 25, 46 (2019). https://doi.org/10.1007/s00894-018-3914-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-018-3914-2