
Abstract
Ionic hydrocarbon compounds that contain hypercarbon atoms, which bond to five or more atoms, are important intermediates in chemical synthesis and may also find applications in hydrogen storage. Extensive investigations have identified hydrocarbon compounds that contain a five- or six-coordinated hypercarbon atom, such as the pentagonal-pyramidal hexamethylbenzene, C6(CH3)62+, in which a hexacoordinate carbon atom is involved. It remains challenging to search for further higher-coordinated carbon in ionic hydrocarbon compounds, such as seven- and eight-coordinated carbon. Here, we report ab initio density functional calculations that show a stable 3D hexagonal-pyramidal configuration of tropylium trication, (C7H7)3+, in which a heptacoordinate carbon atom is involved. We show that this tropylium trication is stable against deprotonation, dissociation, and structural deformation. In contrast, the pyramidal configurations of ionic C8H8 compounds, which would contain an octacoordinate carbon atom, are unstable. These results provide insights for developing new molecular structures containing hypercarbon atoms, which may have potential applications in chemical synthesis and in hydrogen storage.

Possible structural transformations of stable configurations of (C7H7)3+, which may result in the formation of the pyramidal structure that involves a heptacoordinate hypercarbon atom.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neutral carbon compounds involve carbon with the coordination number up to four (tetravalency). Ionic hydrocarbon compounds that contain hypercarbon (or hypercoordiated carbon) atoms, which bond to five or more other atoms, and the related species are a significant feature and important intermediates in various branches of chemistry [1, 2]. Since the 1950s, it has been shown that ionic carbon compounds may contain five- and six-coordinated carbon atoms [2]. These hypercoordiated carbon structures may find valuable applications in hydrogen storage due to a large hydrogen/carbon ratio [3, 4].
Among the ionic hydrocarbons, the methanium ion CH5+, pyramidal (CH)5+, and (CH3)2C5H3+ involve a pentacoordinate carbon atom. These monocations have been observed experimentally [5,6,7,8,9,10,11,12,13,14]. The dications CH62+ and pyramidal (C6H6)2+ contain a hexacoordinate carbon atom, with the latter one recently being confirmed experimentally [1, 5, 15,16,17]. The six-coordinated carbocation, diprotonated methane (CH62+), were investigated with ab initio calculations, which showed, however, contradictory results — the structure was predicted to be either stable [18, 19] or unstable [20]. In addition, the pentagonal-pyramidal hexamethylbenzene, C6(CH3)62+, in which a carbon atom is six-coordinated, was first synthesized in highly acidic conditions at low temperature [5] and recently confirmed with the determination of the corresponding crystal structure [21, 22]. The trication, triprotonated ethane (C2H93+), and the tetracation, tetraprotonated ethane (C2H104+), were also studied with ab initio calculations [23]. The results showed that the stable configurations involve five- and six-coordinated carbon atoms, however, neither structure has been verified experimentally.
The search for ionic hydrocarbon compounds that contain a heptacoordinate or octacoordinate carbon has not yet resulted in any structures confirmed by experiments. Earlier ab initio calculations for the proposed triprotonated methane, the parent heptacoordinate carboncation, (CH7)3+, showed that the trication was a local minimum-energy configuration and its deprotonation was highly exothermic, making it difficult to be prepared experimentally [24]. Similar calculations further suggested that the structure was unstable [23]. In addition, Hogeveen and Kwant suggested a pyramidal structure of the trication C7(CH3)73+ involving seven-coordinated carbon several decades ago [5], but no further theoretical and experimental studies have been done since then, and the possibility of formation of heptacoordinate carbon in C7(CH3)73+ still remains open.
Motivated by possible applications in chemical synthesis and hydrogen storage as well as by the scientific curiosity of the possibility of seven or eight chemical bonds of carbon, we performed ab initio density functional theory (DFT) calculations, through which we explore possible heptacoordinate carbon and octacoordinate carbon in ionic hydrocarbon compounds of C7H7, C8H8, and C7(CH3)7. In particular, we report the identification of a stable three-dimensional pyramidal configuration of (C7H7)3+ that involves a seven-coordinate carbon, suggesting the possibility of heptacoordinate carbon in ionic hydrocarbon compounds. We show that this tropylium trication is stable against deprotonation and dissociation. Our calculations also show that a previously proposed pyramidal trication, C7(CH3)73+, which would involve heptacoordinate carbon, is an unstable configuration. In addition, we find that the pyramidal configurations of ionic C8H8 compounds, which would contain an octacoordinate carbon atom, are unstable.
Methods
The ab initio density functional theory (DFT) calculations were performed using the Vienna Ab Initio Simulation Package [25]. Each hydrocarbon compound under this investigation was placed into a large supercell with the size of 20 × 20 × 20 Å to simulate isolated compounds [26]. Convergence was checked with a larger supercell, 30 × 30 × 30 Å, which showed insignificant changes in results. The Perdew-Burke-Ernzerhof generalized gradient approximations [27] for the exchange-correlation functionals were employed in the DFT calculations, and the electron-core interactions were treated with the projector augmented wave (PAW) method [28, 29]. Plane-wave basis sets with a cut-off energy of 500 eV and one sampling k point at Gamma in the three-dimensional Brillouin zone of the supercell were used in the calculations. Convergence check was performed with higher cut-off energies (up to 600 eV) and more k points (up to 8 k points). Optimization of the structure was performed for each supercell via a conjugate-gradient technique using the total energy and the Hellmann-Feynman forces on the atoms [26], and all configurations were optimized until the forces on each atom were smaller than 0.03 eV/Å. DFT calculations, with the climbing image nudged elastic band method [30], were used to determine the energy barriers when structural transformations were considered. Charges of individual atoms were quantitatively determined using Bader charge analysis [31, 32].
Results and discussion
We started with tropylium monocation (or cycloheptatrienylium monocation), which has a formula of (C7H7)+ or (CH)7+, the stable structure of which is experimentally observed to be planar, as shown in Fig. 1a and b [33]. In this 2D structure, seven carbon atoms form a planar ring with each carbon bonded to three other atoms. Hence, each carbon atom in planar (C7H7)+ is three-coordinated. The optimized structure, determined by our ab initio DFT calculation, shows that the C-C and C-H bonds have bond lengths of 1.40 Å and 1.09 Å, respectively.

DFT-optimized configurations of (C7H7)+: (a) Top view of the 2D planar structure; (b) side view of the planar structure; (c) top view of the 3D structure; and (d) side view of the 3D structure. Carbon and hydrogen atoms are represented by larger and smaller balls, respectively
The corresponding 3D configuration, as shown in Fig. 1c and d, was obtained after optimization of the initial pyramidal configuration. We find that the pyramidal structure for (C7H7)+ is not a stable configuration, and optimization of the pyramidal structure leads to the local minimum-energy 3D configuration shown in Fig. 1c and d. However, the six-membered carbon ring does not hold on a 2D plane, resulting in the configuration with a pentacoordinated carbon atom. This 3D (C7H7)+ configuration is much higher in total energy, by 546 kJ mol-1, than planar (C7H7)+, indicating that the 3D structure with pentacoordinate carbon is very unlikely to occur.
Similar to (C7H7)+, the pyramidal structure of tropylium dication, (C7H7)2+ is unstable. The optimized 3D configuration from the initial pyramidal structure is very similar to that of (C7H7)+ and involves pentacoordinate carbon (not shown here). The planar configuration of (C7H7)2+ was also optimized, and the 2D ring structure was found to be stable. The geometry of the 2D ring structure of (C7H7)2+ is almost the same as that of (C7H7)+ (Fig. 1a and b), but the C-C and C-H bond lengths of 1.42 Å and 1.10 Å for (C7H7)2+ are slightly longer than the corresponding bond lengths for (C7H7)+. The 3D configuration is much higher in total energy than the planar structure (by 284 kJ mol-1).
Unlike (C7H7)+, however, there exists another stable configuration for (C7H7)2+, as shown in Fig. 2. This configuration is slightly different from the 2D ring structure as one of the CH moieties is slightly off the ring plane (as such, we name the structure as an off-ring configuration), and its total energy is lower, by 8 kJ mol-1, than that of the 2D ring structure. The very small difference in total energy indicates that these two structures (2D planar and off-ring) can co-exist.

DFT-calculated off-ring configuration for (C7H7)2+: (a) Top view; and (b) side view
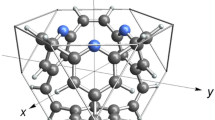
On the other hand, tropylium trication, (C7H7)3+, shows more distinct properties than the corresponding monocation and dication. The 3D pyramidal structure of (C7H7)3+, as shown in Fig. 3, in which a heptacoordinate carbon atom is involved, is a stable configuration. Optimization of the configuration does not lead to any change of the pyramidal shape and the heptacoordinate carbon remains intact. Furthermore, the total energy of the pyramidal configuration of (C7H7)3+ is lower than that of the corresponding 2D ring configuration. The total energy difference is small (11 kJ mol-1), but it demonstrates that the pyramidal configuration is energetically more stable than the planar structure.

DFT-optimized 3D pyramidal configuration for (C7H7)3+: (a) Top view; and (b) side view
The base of the pyramidal configuration is formed by a six-membered carbon ring with each carbon bonded to a hydrogen atom. The C-C and C-H bond lengths are determined to be 1.44 Å and 1.11 Å, respectively. The carbon atom at the top vertex of the hexagonal pyramid is bonded to all six carbons on the base, forming six C-C bonds each with a bond length of approximately 1.82 Å. The apical carbon is also bonded to a hydrogen atom, and the C-H bond length is 1.12 Å. Therefore, the apical carbon is bonded to seven neighboring atoms, making it heptacoordinated.
As a comparison, the optimized structure of the two-dimensional ring configuration of (C7H7)3+ was also determined. The configuration is almost the same as those for the monocation (C7H7)+ (Fig. 1a and b) and dication (C7H7)2+, except for the fact that the C-C bonds (with bond lengths of 1.45 Å) in planar (C7H7)3+ are longer than those in planar (C7H7)+ and (C7H7)2+.
Similar to (C7H7)2+, there exists a stable off-ring configuration for (C7H7)3+, as shown in Fig. 4. This configuration involves an off-plane C-H bond, which is almost vertical to the plane formed with other carbon atoms. The total energy of the off-ring configuration is lower than those of the 3D pyramidal and the 2D ring structures by 25 kJ mol-1 and 36 kJ mol-1, respectively. Therefore, the off-ring structure is energetically the most stable configuration for (C7H7)3+, while both 3D pyramidal and the 2D ring structures are metastable.

DFT-calculated off-ring configuration for (C7H7)3+: (a) Top view; and (b) side view
As the 3D pyramidal configuration that involves heptacoordinate carbon is energetically metastable, we further investigated its stability against the structural transformation to the off-ring configuration. We determined the optimal transition pathway and the corresponding energy barrier for this structural transformation. Figure S1 shows the configuration for the transition state, in which the CH species being transferred from the top of the pyramidal configuration is located almost on the top of the bridge site of two carbon atoms in the carbon ring.
The energy barrier was calculated to be 38 kJ mol-1 (Fig. 5). Because of this activation barrier, the metastable pyramidal configuration may not undergo a structural transformation, especially under the condition of low temperatures. This case is similar to the pyramidal hexamethylbenzene, C6(CH3)62+, in which hexacoordinate carbon is involved. We calculated the optimized structures of the pyramidal, flat ring, and off-ring configurations for C6(CH3)62+ (Fig. S2). The pyramidal configuration has a lower total-energy than the 2D ring structure (by 29 kJ mol-1), consistent with the result reported by Malischewski et al. [21, 34]. However, the off-ring configuration has an even lower total energy than the pyramidal configuration (by 7 kJ mol-1), indicating that pyramidal configuration is metastable. Nevertheless, the metastable pyramidal configuration has been experimentally observed recently [21].

Diagram of the energy differences and the activation energies for the structural transformations between the 2D ring, off-ring, and 3D pyramidal configurations. TS-1 and TS-2 refer to the transition states
Furthermore, as a comparison, we calculated the energy barrier for the structural transformation of the 2D ring configuration of (C7H7)3+ to the off-ring configuration, and we obtained a value of 2 kJ mol-1 (Fig. 5). This small energy barrier suggests that, even at low temperatures, the 2D ring configuration may transfer to the off-ring configuration.
We have also calculated the change of the total energy during the process of deprotonation and dissociation of the CH+ species. The energy cost of both processes are very high: 332 kJ mol-1 when the hydrogen at the top of the pyramid is separated from the rest, 377 kJ mol-1 if a hydrogen bonded to a carbon atom at the pyramid base is separated, and 514 kJ mol-1 when the CH+ species is dissociated from the (C6H6)2+ ring, indicating that the pyramidal configuration is highly stable against deprotonation and dissociation.
Our computational results for tropylium ions are consistent with Hückel’s rule (the 4n + 2 rule) [35, 36] for 2D aromaticity and the 4n + 2 interstitial electron rule for 3D aromaticity [37, 38]. The tropylium monocation (C7H7)+ that takes a 2D ring structure is an aromatic species. There are 6 π-electrons in the ring because of the monocation nature, and Hückel’s rule is hence satisfied (4n + 2, and n = 1) [35,36,37]. On the other hand, the stable trication (C7H7)3+ takes the 3D pyramidal configuration. This configuration can be regarded as a combination of dication (C6H6)2+ and monocation (CH)+, which provides 4 and 2 electrons, respectively, for the bonding between them. Therefore, there are 6 valence electrons participating in the bonding between the apical carbon atom and the six carbons on the base of the pyramid, and the 4n + 2 (n = 1) interstitial electron rule for 3D aromaticity is satisfied [37].
While the bonding of stable 3D aromatic species obeys the 4n + 2 interstitial electron rule, the reverse statement may not be true. That is, a satisfaction of the 4n + 2 interstitial electron rule does not necessarily lead to a stable 3D aromatic species. To elucidate this point, we examined the case of the hexagonal-pyramidal heptamethylbenzene, C7(CH3)73+, which was first proposed by Hogeveen and Kwart as a possible ionic hydrocarbon compound that would involve a heptacoordinate carbon [5]. The configuration can be viewed as a bonded structure of C(CH3)+ and planar C6(CH3)62+, as shown in Fig. 6a. The six-membered carbon ring of the base contributes 4 interstitial electrons, while C(CH3)+ provides 2, making the total number of the interstitial electrons 6, which satisfies the 4n + 2 (n = 1) interstitial electron rule. However, our ab initio calculations show that the pyramidal configuration is unstable. Optimization of the structure results in a structural change (Fig. 6b) — the pyramidal configuration and the involved heptacoordinate are no longer kept. This result is different from the case of pentagonal-pyramidal hexamethylbenzene, C6(CH3)62+, which obeys the 4n + 2 (n = 1) interstitial electron rule and the structure is stable [21].

Schematics of the configurations of C7(CH3)73+: (a) The proposed pyramidal structure (unstable); and (b) the structure after the pyramidal configuration is fully optimized. Only side views are shown
It becomes intriguing to explore octacoordinate carbon using the same chemical principle. We examined possible octacoordinate carbon in the configurations of mono-, di-, tri-, and tetra-cations of C8H8, which are (C8H8)+, (C8H8)2+, (C8H8)3+, and (C8H8)4+, respectively. The optimized equilibrium configurations are shown in Figs. S3–S8.
(C8H8)+, (C8H8)2+, (C8H8)3+, and (C8H8)4+ have a stable 2D ring configuration. The pyramidal configurations of both (C8H8)+ and (C8H8)2+ are unstable, and they spontaneously transferred to the configurations that have much higher energies than the corresponding 2D ring structures, by 89 kJ mol-1 and 229 kJ mol-1, respectively, indicating that the 2D ring structures are much more likely to occur.
Instead, we find that the pyramidal configurations for (C8H8)3+ and (C8H8)4+ are stable, but both have a much higher energy than the corresponding 2D ring configurations, by 290 kJ mol-1 and 145 kJ mol-1, respectively, implying that the pyramidal configurations are unlikely to occur. In addition, the off-ring configurations for (C8H8)3+ and (C8H8)4+ have a much lower energy than the corresponding pyramidal configurations by 196 kJ mol-1 and 162 kJ mol-1, respectively. The energy barriers for the pyramidal configurations to transfer to the off-ring configurations are determined to be very small (10 kJ mol-1 and 6 kJ mol-1 for (C8H8)3+ and (C8H8)4+, respectively). Therefore, the pyramidal configurations are kinetically unstable at room temperature.
Conclusions
In conclusion, ab initio DFT calculations reported here demonstrate that heptacoordinate carbon is possible in the ionic hydrocarbon compound tropylium trication (C7H7)3+. The 3D hexagonal-pyramidal (C7H7)3+, which contains a heptacoordinate carbon atom, is predicted to be a metastable configuration. It is highly stable against dissociation of CH+ and deprotonation, and fairly stable against structural transformation. The result can be explained by the 4n + 2 interstitial electron rule for 3D aromaticity. On the other hand, tropylium monocation and dication, (C7H7)+ and (C7H7)2+, as well as the previously proposed hexagonal-pyramidal heptamethylbenzene, C7(CH3)73+, do not involve any heptacoordinate carbon. In addition, none of the ionic C8H8 would involve octacoordinate carbon, and the proposed heptagonal-pyramidal configurations are all shown to be unstable. These results provide insights for developing new molecular structures containing hypercarbon atoms, which may have potential applications in chemical synthesis and in hydrogen storage.
References
Olah GA (1995) Angew Chem Int Ed 34:1393–1405
Olah GA, Prakash G, Wade K, Molnár Á, Williams RE (2011) Hypercarbon chemistry, 2nd edn. Wiley, Hoboken, p 85–147
Du J, Sun X, Jiang G, Zhang C (2016) Int J Hydrog Energy 41:11301–11307
Gao Y, Shao N, Zhou RL, Zhang GL, Zeng XC (2012) J Phys Chem Lett 3:2264–2268
Hogeveen H, Kwant PW (1975) Acc Chem Res 8:413–420
Surya Prakash G, Rasul G, Olah GA, Phys J (1998) J Phys Chem A 102:2579–2583
Olah GA, Rasul G (1997) Acc Chem Res 30:245–250
Boo DW, Lee YT (1995) J Chem Phys 103:520–530
Williams RE (1971) Inorg Chem 10:210–214
Stohrer WD, Hoffmann R (1972) J Am Chem Soc 94:1661–1668
Masamune S, Sakai M, Ona H, Jones AJ (1972) J Am Chem Soc 94:8956–8958
Kemp-Jones AV, Nakamura N, Masamune S (1974) J Chem Soc Chem Commun 109–110
Masamune S, Sakai M, Kemp-Jones A, Ona H, Venot A, Nakashima T (1973) Angew Chem Int Ed 12:769–771
Coates R, Fretz E (1977) Tetrahedron Lett 18:1955–1960
Hogeveen H, Kwant PW (1974) J Am Chem Soc 96:2208–2214
Hogeveen H, Kwant PW (1973) Tetrahedron Lett 14:1665–1670
Jašík J, Gerlich D, Roithová J (2014) J Am Chem Soc 136:2960–2962
Lammertsma K, Barzaghi M, Olah GA, Pople JA, Schleyer P, Simonetta M (1983) J Am Chem Soc 105:5258–5263
Lammertsma K, Olah GA, Barzaghi M, Simonetta M (1982) J Am Chem Soc 104:6851–6852
Jursic BS (1999) J Chem Res Synop :502–503
Malischewski M, Seppelt K (2017) Angew Chem Int Ed 56:368–370
Malischewski M, Seppelt K (2017) Angew Chem Int Ed 56:16495–16497
Rasul G, Prakash GS, Olah GA (2010) J Phys Chem A 114:12124–12127
Olah GA, Rasul G (1996) J Am Chem Soc 118:8503–8504
Kresse G, Furthmuller J (1996) Phys Rev B 54:11169–11186
Payne MC, Teter MP, Allan DC, Arias T, Joannopoulos J (1992) Rev Mod Phys 64:1045
Perdew JP, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:3865–3868
Blochl PE (1994) Phys Rev B 50:17953–17979
Kresse G, Joubert D (1999) Phys Rev B 59:1758–1775
Henkelman G, Uberuaga BP, Jonsson H (2000) J Chem Phys 113:9901–9904
Bader RFW (1990) Atoms in molecules - a quantum theory. Clarendon, Oxford
Henkelman G, Arnaldsson A, Jónsson H (2006) Comput Mater Sci 36:354–360
Von W, Doering E, Knox L (1957) J Am Chem Soc 79:352–356
Klein JEMN, Havenith RWA, Knizia G (2018) Chem Eur J. https://doi.org/10.1002/chem.201705812
Hückel E (1931) Z Phys 70:204–286
Roberts JD, Streitwieser Jr A, Regan CM (1952) J Am Chem Soc 74:4579–4582
Jemmis ED, Schleyer P v R (1982) J Am Chem Soc 104:4781–4788
McKee WC, Agarwal J, Schaefer HF, Schleyer P v R (2014) Angew Chem Int Ed 53:7875–7878
Acknowledgments
The authors would like to thank Michael Lee from the Oklahoma School of Science and Mathematics (now at the University of Oklahoma) for valuable discussions at the early stage of this work. The calculations have been performed using computational resources at the OU Supercomputing Center for Education & Research (OSCER) at the University of Oklahoma.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(DOCX 1941 kb)
Rights and permissions
About this article

Cite this article
Wang, G., Rahman, A.K.F. & Wang, B. Ab initio calculations of ionic hydrocarbon compounds with heptacoordinate carbon. J Mol Model 24, 116 (2018). https://doi.org/10.1007/s00894-018-3640-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-018-3640-9