
Abstract
The structures and energetics of two dihydrochalcones (phloretin and its glycoside phlorizin) were examined with density functional theory, using the B3LYP, M06-2X, and LC-ω PBE functionals with both the 6-311G(d,p) and 6-311 + G(d,p) basis sets. Properties connected to antioxidant activity, i.e., bond dissociation enthalpies (BDEs) for OH groups and ionization potentials (IPs), were computed in a variety of environments including the gas-phase, n-hexane, ethanol, methanol, and water. The smallest BDEs among the four OH groups for phloretin (three for phlorizin) were determined (using B3LYP/6-311 + G(d,p) in water) to be 79.36 kcal/mol for phloretin and 79.98 kcal/mol for phlorizin while the IPs (at the same level of theory) were obtained as 139.48 and 138.98 kcal/mol, respectively. By comparing with known antioxidants, these values for the BDEs indicate both phloretin and phlorizin show promise for antioxidant activity. In addition, the presence of the sugar moiety has a moderate (0-6 kcal/mol depending on functional) effect on the BDEs for all OH groups. Interestingly, the BDEs suggest that (depending on the functional chosen) the sugar moiety can lead to an increase, decrease, or no change in the antioxidant activity. Therefore, further experimental tests are encouraged to understand the substituent effect on the BDEs for phloretin and to help determine the most appropriate functional to probe BDEs for dihydrochalcones.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
There is considerable interest in studying polyphenols, and thus, experimental and computational studies on the isolation/extraction, synthesis, characterization, and reactivity of such compounds are prevalent in the very recent literature [1,2,3,4,5,6,7,8,9,10]. Dihydrochalcones (DHCs) are a class of polyphenols that are present in extracts of more than forty weed plants and deciduous trees from families such as Leguminoseae, Laureaceae, and Lorantaceae [11]. These compounds attract considerable attention for being used in the human diet as functional foods due to their pharmacological activity, including antioxidant, antibacterial, anti-inflammatory, antitumor, and antiviral properties [12,13,14,15,16]. In terms of antioxidant activity, it is well known that the potential of a given substance can be probed through its capability to scavenge free radicals by (mainly) the mechanisms of hydrogen-atom transfer (HAT) and single electron transfer (SET) [17,18,19,20,21]. In HAT, where an H-atom is transferred to a free radical, i.e.,
there is a strong dependence on the O-H bond dissociation enthalpy (BDE), since there will be higher antioxidant activity when there is a weaker O-H bond [17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37]. The BDE (i.e., energy to break the O-H bond) is computed as the difference in the heat of formation between the molecule (ArOH) and corresponding radical (ArO.)[38]. On the other hand, in SET, a single electron is transferred from the molecule (ArOH) to the free radical (R.), i.e.,
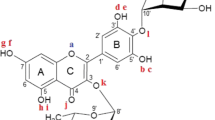
Clearly, the smaller the ionization potential (IP) for ArOH, the lower the energetic cost to abstract an electron. By examining the BDEs and IPs of DHCs, additional information can be obtained about potential compounds with applications as phytotherapeutics [39]. Very recently, we performed a computational study on two flavonols that were isolated from Loranthaceae family plant extracts: kaempferol 3-O-α-L-arabinofuranosyl-(1→3)-α-L-rhamnoside and quercetin 3-O-α-L-arabinofuranosyl-(1→3)-α-L-rhamnoside [40], which are glycosylated versions of kaempferol and quercetin, respectively. One of the goals of the previous study was to probe how much the presence of the sugar group would affect the values of BDEs and IPs. In the present work, we extend this idea to examining phloretin and its glycoside phlorizin. The structures of both compounds can be seen in Fig. 1. Phloretin and phlorizin have been studied as candidate molecules for skin-based drug delivery [41], anti-inflammatories [42], and UV light-induced photodamage protectors [43]. There are various pharmacological studies on phloretin [44,45,46,47], including in vivo and in vitro investigations on the antioxidant activity [48, 49]. Two studies reporting BDEs for phloretin have previously been published: Kozlowski et al. [50] computed BDEs for a series of chalcones and dihydrochalcones in the gas-phase, methanol, and water using density functional theory (DFT) with the B3P86 functional and the 6-31 + G(d,p) basis set. Yan et al. [51] utilized the B3LYP functional [52,53,54,55] and the 6-311G(d,p) basis set [56,57,58,59] to compute the BDEs for phloretin in the gas-phase, benzene, and water; no IPs were determined. To the best of our knowledge, no investigation on BDEs or IPs for phlorizin were performed to date. Hence, the present work provides additional information on the potential antioxidant activity of these compounds including the role of the sugar. The choice of functional and basis set on the effect of the sugar has been examined to determine that a consistent story emerges independent of the computational method. The geometries of phloretin and its glycoside phlorizin were optimized to obtain structural parameters, and then based on these structures, the corresponding BDEs and IPs were determined in the gas-phase, n-hexane, ethanol, methanol, and water. A comparison of the present values to previous computational and experimental results for other flavonols suggest both compounds should exhibit antioxidant activity. In addition, the presence of the sugar moiety has only a modest effect on the BDEs and IPs. Therefore, experimental investigations on BDEs of both compounds are encouraged to verify the predictions made in the present work.

Representation of the chemical structures, along with atom numbering, for the compounds of interest in the present work
Computational methods
DFT was the computational approach applied in the present work. In a very recent study, La Rocca et al. [60] benchmarked twenty-one (21) commonly used exchange-correlation functionals for the determination of the BDEs and IPs for two selected molecules that are well known to present antioxidant activity: quercetin and edaravone. The conclusion was that M05-2X [61], M06-2X [62], and LC-ω PBE [63] were the preferred functionals to compute the antioxidant behavior. Hence, in the present work, we decided to use one of Minnesota family functionals (M06-2X), LC-ω PBE and the widely used B3LYP (to directly compare our results with BDEs for phloretin avaliable in the literature). Geometry optimization (using default convergence criteria and without symmetry constraints) of the neutral molecules and their radicals were carried out using the B3LYP, M06-2X, and LC-ω PBE exchange-correlation functionals [52,53,54,55, 62, 63] with both the 6-311G(d,p) and 6-311 + G(d,p) basis sets [56,57,58,59]. The initial structure for the optimization of the neutral phloretin was built following the results achieved in the conformational analysis performed by Kozlowski et al. [50]. In terms of phlorizin, the structure used was based on the crystal structure presented by Aufmkolk et al. [64]. Additionally, computations on the energies of other conformers were performed to verify if the crystallographic data corresponded to the lowest energy conformer for the given computational method. Regarding the differences, there are several low-lying conformers that are very close in energy (0.001–0.4 kcal/mol). Therefore, these are clearly energetically accessible; a figure with the details of the structural parameters of the conformer utilized can be found in the Supplementary Information. For the open-shell species, the unrestricted formalism (i.e., UB3LYP, UM06-2X, U-LC-ω PBE) was used for all computations. To confirm the conformations as minima, to evaluate the zero-point energy (ZPE) corrections, and to determine enthalpies (at 298 K), vibrational frequencies were computed for all optimized structures. Solvent effects were included using the integral equation formalism polarizable continuuum model (IEF-PCM; which shall hereafter be simply referred to as PCM) [65,66,67]. The solvents considered included n-hexane (𝜖= 1.8819), ethanol (𝜖= 24.852), methanol (𝜖= 32.613), and water (𝜖= 78.3553). Single-point second order Møller-Plesset perturbation theory computations, MP2/6-31G(d,p)//B3LYP/6-311 + G(d,p), were also performed to provide comparison between the results obtained using a wave function based method with those obtained with DFT.
The BDEs were determined using the enthalpies of formation for the radical generated via H-atom abstraction H f (ArO.), for the H-atom H f (H.), and for the neutral molecule H f (ArOH), such that
For each compound, the IPs were determined as the difference between the (electronic + ZPE) energies for the neutral (ArOH) and the cation (ArOH+). All computations in the work presented here were accomplished using the Gaussian 09 software suite [68].
Results and discussion
Structures
Table 1 presents selected optimized bond lengths, bond angles, and dihedral angles for phloretin and phlorizin determined in the gas-phase using the B3LYP, M06-2X, and LC-ω PBE functionals with the 6-311G(d,p) and 6-311 + G(d,p) basis sets. For both compounds, the bond lengths and bond angles agree very well with each other (within 0.003 Å and 1 degree) for a given functional when the basis set is changed (as expected); the dihedrals for phlorizin show a larger (up to 15 degrees) basis set dependence. Thus, for the B3LYP, M06-2X, and LC-ω PBE approaches, there is only a very weak dependence when describing the bond lengths and angles for the two compounds of interest. This is in agreement to what was reported in previous studies on other phenolic compounds [25, 40].
The present results (determined in all the approaches used) suggest there is a moderate interaction between H8 and O a in phloretin (with the distance between H8 and O a being approximately 1.6 Å). Hence, the \(r_{(\mathrm {H}_{8}-\mathrm {O}_{7})}\) distance is slightly larger than any other r(H−O) bond for this compound. This behavior was also observed in previous studies for other molecules containing a hydroxyl neighboring the carbonyl moiety [39, 40]. In phlorizin, the presence of the galactose causes an enlargement in the \(r_{(\mathrm {C}_{2^{\prime }}-\mathrm {O}_{7})}\) distance and a small change in the angle \(\theta _{(C_{1^{\prime }}-C_{2^{\prime }}-O_{7})}\). In addition, the torsion angles \(\theta _{(C_{\alpha }-C_{\beta }-C_{1}-C_{2})}\), \(\theta _{(C_{\alpha }-C_{\beta ^{\prime }}-C_{1^{\prime }}-C_{2^{\prime }})}\), and \(\theta _{(C_{\alpha }-C_{\beta }-C_{1^{\prime }}-C_{6^{\prime }})}\) experience major changes due to the sugar moiety, which indicates the loss of coplanarity between ring A and the keto group. The rotation around \(\theta _{(C_{\alpha }-C_{\beta ^{\prime }}-C_{1^{\prime }}-C_{2^{\prime }})}\) can have a significant impact on the antioxidant activity of phlorizin.
Determination of BDEs and IPs
Tables 2 and 3 show the BDEs computed at the B3LYP/6- 311 + G(d,p), M06-2X/6-311 + G(d,p), and LC-ω PBE/6-311 + G(d,p) levels of theory in the gas-phase, n-hexane, ethanol, methanol, and water for phloretin and phlorizin, respectively. Corresponding DFT results with the 6-311G(d,p) basis set are provided in the Supplementary Material. MP2/6-31G(d,p)//B3LYP/6-311 + G(d,p) results in the gas-phase and water are included for comparison; however, as has been noted previously [69]. MP2 overbinds when compared to DFT and experimental results. In general, polar solvents (such as ethanol, methanol, and water) are the most important ones for experimentally testing antioxidant activity. However, in a previous study by our group [40], we computed BDEs in n-hexane and, thus, this solvent was included in the present work to compare the behavior for phloretin and phlorizin. For phloretin, results from Yan et al. [51] determined at the B3LYP/6-311G(d,p) level of theory in the gas-phase and water and results from Kozlowski et al. [50] computed at the B3P86/6-31 + G(d,p) level of theory in the gas-phase, methanol, and water are also included in Table 2 for comparison purposes.
A very small basis set dependence (0.01-1 kcal/mol) can be observed for the BDEs computed in the gas-phase. In addition, most of the BDEs obtained exhibit values where 6-311 + G(d,p)< 6-311G(d,p) using B3LYP, M06-2X and LC-ω PBE for both phloretin and phlorizin.
In both compounds, the computationally determined BDEs using DFT (in gas phase) for the majority of the OH groups have values lower than 91.98 kcal/mol, that is the BDE determined previously [70] for gallic acid at B3LYP/6-31G(d,p) in the gas phase. Additionally, the value of 90.93 kcal/mol for gallic acid (in water) is also higher than most of BDEs computed for phloretin and phlorizin in the same solvent environment. These observations suggest that both compounds studied in this work are promising in presenting antioxidant activity, given that gallic acid is one of the standards in experimental tests of antioxidant activity [71,72,73] and present a common structural moiety with phloretin and phlorizin.
For both phloretin and phlorizin (for all the functionals and basis sets and in all the solvents), the relative BDEs for the OH groups are 4-ArOH < 6′-ArOH < 4′-ArOH < 2′-ArOH. A slightly different trend (6′-ArOH < 4-ArOH) was observed in the results of Yan et al. [51] for phloretin; however, the difference between the two BDEs is less than 2 kcal/mol and Yan et al.’s values exhibit differences with the present BDE values at the same level of theory. This discrepancy is due to the use of a different conformer of phloretin by those authors as well as (perhaps) subtle differences in the computational methodology. Yan et al. [51] utilized the GAMESS [74] software package (hence different convergence thresholds and definition of B3LYP) and a different solvation model. Interestingly, the MP2/6-31G(d,p)//B3LYP/6-311 + G(d,p) results for the present conformer also show the BDE for 6′-ArOH to be lower than that for 4-ArOH but these results are strongly overbound compared to the DFT ones as has been observed previously [69]; hence, the primary discussions focus on the DFT-determined BDEs. On the other hand, BDEs computed by Kozlowski et al. [50] are comparable to the ones obtained in the present work. Our BDEs computed using the B3LYP functional are shifted by approximately 4 kcal/mol when compared to the ones determined using the B3P86 functional; in agreement to what is stated by Kozlowski et al. [50] with regard to relative energies determined with these two functionals. 2′-ArOH is well known for presenting the highest BDE due to the occurrence of the hydrogen bonding interaction between the H atom from hydroxyl and the O atom from the keto group already discussed in the previous section. The inclusion of the galactose causes the following effects on the values of BDEs: results obtained from the LC-ω PBE/6-311 + G(d,p) approach in the gas-phase indicate that the sugar moiety would cause a decrease of 1.22 kcal/mol in the 4-ArOH BDE which is in agreement with the behavior observed for kaempferol 3-O-α-L-arabinofuranosyl-(1→3)-α-L-rhamnoside and quercetin 3-O-α-L-arabinofuranosyl-(1→3)-α-L-rhamnoside in a recent study of our research group [40]. The decrease is also observed for computations taking into account the solvents n-hexane, ethanol, and methanol; however, in smaller fashions. The 4-ArOH BDEs determined at the B3LYP/6-311 + G(d,p) level of theory in methanol and n-hexane for phlorizin also present a decrease (of approximately 1.16 kcal/mol) when compared to the ones computed at the same level for phloretin. In the case of employing M06-2X/6-311 + G(d,p), the BDEs of the 4-ArOH are slightly higher for phlorizin than the ones for phloretin. The BDE values of 6′-ArOH and 4′-ArOH are changed (sometimes increasing a small amount and other times having a significant decrease) with the sugar inclusion. Interestingly, the presence of the galactose causes different effects on all the BDE values of 6′-ArOH and 4′-ArOH, respectively. For instance, at the M06-2X/6-311 + G(d,p) level of theory the BDEs of the 6′-ArOH barely increase while the BDEs of the 4′-ArOH decrease up to 5.75% in their values when the sugar moiety is present. This can be attributed to the rotation of the A ring (see torsion angles and \(\theta _{(C_{\alpha }-C_{\beta }-C_{1^{\prime }}-C_{6^{\prime }})}\)) that can lead to a sterically more (or less) crowded chemical site depending on the configuration established by the compound.
There are previous investigations reporting that the inclusion of a sugar moiety in a polyphenol increases the BDEs [36, 37]. In the previous study we performed on kaempferol 3-O-α-L-arabinofuranosyl-(1→3)-α-L-rhamnoside and quercetin 3-O-α-L-arabinofuranosyl-(1→3)-α-L-rhamnoside [40], which are glycosylated versions of kaempferol and quercetin, respectively, we observed a small decrease and/or increase in the BDEs (particularly for quercetin 3-O-α-L-arabinofuranosyl-(1→3)-α-L-rhamnoside) when comparing to the ones of the parent molecules. In the present study, as seen in Tables 2 and 3, the BDE values for phlorizin (4-ArOH) are comparable (sometimes slightly lower or slightly higher) than those computed for phloretin (6′-ArOH). Thus, the presence of the sugar substituent can lead to an increase (or decrease) in the antioxidant activity of phlorizin when in comparison to phloretin which is in agreement with our previous study [40].
The IPs for phloretin and phlorizin computed using B3LYP, M06-2X, and LC-ω PBE with the 6-311G(d,p) and 6-311 + G(d,p) basis sets in the gas-phase, n-hexane, ethanol, methanol, and water are presented in Tables 4 and 5, respectively. Again, MP2/6-31G(d,p)//B3LYP/6-311 + G(d,p) results are included. In general, it is possible to notice a considerable decrease in the IPs for both phloretin and phlorizin when in the presence of polar solvents (i.e., ethanol, methanol, and water) than for non-polar n-hexane. As reported in several previous studies [17,18,19], the surrounding environment has a strong impact on the IP values, and thus, the polar solvent facilitates the SET mechanism (related to the IP values) by providing charge separation. The IPs of phlorizin are always lower than those of phloretin at B3LYP with 6-311-G(d,p) and 6-311 + G(d,p) basis sets, which is in agreement to what was observed in our recent work for other sugar substituted compounds [40]. Using M06-2X and LC-ω PBE, the pattern is not the same; in some solvents phlorizin has higher IPs (in others, lower IPs) than phloretin. In water, B3LYP predicts phloretin to have a lower IP than phlorizin, while M06-2X and LC-ω PBE both predict the reverse trend. Thus, regarding the SET mechanism the presence of the sugar moiety should have a minor influence, but the nature of the influence (positive or negative) cannot be unambiguously assigned from the present computations.
Summary
The parent molecule phloretin and its glycosylated version phlorizin, see Fig. 1, have been examined computationally utilizing DFT with the B3LYP, M06-2X, and LC-ω PBE functionals and with both the 6-311G(d,p) and 6-311 + G(d,p) basis sets. The focus of the investigation has been on the structural and energetic parameters including both BDEs and IPs, which provide information on the potential antioxidant activities. As the values of BDEs are considerably lower than the ones probed for IPs (in the gas phase or in any given solvent environment), the HAT mechanism is preferred over the SET mechanism. The BDEs determined suggest both phloretin and phlorizin hold promise for exhibiting antioxidant activity, especially phlorizin. Interestingly, the present BDE results suggest that (depending of the functional chosen) the sugar moiety can lead to an increase, decrease, or no change in the antioxidant activity. Hence, further experimental tests are encouraged in order to mitigate the substituent effect on phloretin and test the best choice of functional to probe BDEs for DHCs.
Supplementary Information
Cartesian coordinates for the optimized geometries determined using the B3LYP, M06-2X, LC-ω PBE functionals and the 6-311 + G(d,p) basis set in the gas-phase, n-hexane, ethanol, methanol, and water, can be found in the Supplementary Materials.
References
Fernandez-Pastor I, Fernandez-Hernandez A, Rivas F, Martinez A, Garcia-Granados A, Parra A (2016) Synthesis and antioxidant activity of hydroxytyrosol Alkyl-Carbonate derivatives. J Nat Prod 79:1737–1745
Wu G, Johnson SK, Bornman JF, Bennett SJ, Fang Z (2017) Changes in whole grain polyphenols and antioxidant activity of six sorghum genotypes under different irrigation treatments. Food Chem 214:199–207
Murthy PK, Mary YS, Suneetha V, Panicker CY, Armaković S, Armaković SJ, Giri L, Suchetan PA, Alsenoy CV (2017) Towards the new heterocycle based molecule: synthesis, characterization and reactivity study. J Mol Struct 1137:589–605
Varnali T (2016) A comparison of scytonemin and its carbon analogue in terms of antioxidant properties through free radical mechanisms and conformational analysis: a DFT investigation. J Mol Model 22:213
Grajeda-Iglesias C, Figueroa-Espinoza MC, Barouh N, Baréa B, Fernandes A, de Freitas V, Salas E (2016) Isolation and characterization of anthocyanins from hibiscus sabdariffa flowers. J Nat Prod 79:1709–1718
Okumura K, Hosoya T, Kawarazaki K, Izawa N, Kumazawa S (2016) Antioxidant activity of phenolic compounds from fava bean sprouts. J Food Sci 81:1394–1398
Benzon KB, Sheena MY, Panicker CY, Armaković S, Armaković SJ, Pradhan K, Nanda AK, Alsenoy CV (2017) Studies on the synthesis, spectroscopic analysis, molecular docking and DFT calculations on 1-hydroxy-2-(4-hydroxyphenyl)-4,5-dimethyl-imidazol 3-oxide. J Mol Struct 1130:644–658
Ranjith PK, Mary YS, Panicker CY, Anto PL, Armaković S, Armaković SJ, Musiol R, Jampilek J, Alsenoy CJ (2017) New quinolone derivative: spectroscopic characterization and reactivity study by DFT and MD approaches. J Mol Struct 1135:1–14
Apak R, Ozyurek M, Guclu K, Capanoglu E (2016) Antioxidant activity/capacity measurement. 1. Classification, physicochemical principles, mechanisms, and electron transfer (ET)-based assays. J Afric Food Chem 64:997–1027
Malig TC, Ashkin MR, Burman AL, Barday M, Heyne BJ, Back TG (2017) Comparison of free-radical inhibiting antioxidant properties of carvedilol and its phenolic metabolites. Med Chem Comm 8:606–615
Rahman A-U (2016) Studies in natural products chemistry, vol 51. Elsevier, Oxford, pp 253–373
Aswathy VV, Alper-Hayta S, Yalcin G, Mary YS, Panicker CY, Jojo PJ, Kaynak-Onurdag F, Armaković S, Armaković SJ, Yildiz I, Alsenoy CV (2017) Modification of benzoxazole derivative by bromine-spectroscopic, antibacterial and reactivity study using experimental and theoretical procedures. J Mol Struct 1141:495–511
Plaza M, Kariuki J, Turner C (2014) Quantification of individual phenolic compounds’ contribution to antioxidant capacity in apple: a novel analytical tool based on liquid chromatography with diode array, electrochemical, and charged aerosol detection. J Agric Food Chem 62:409–418
Zhang XC, Chen F, Wang MF (2014) Antioxidant and antiglycation activity of selected dietary polyphenols in a cookie model. J Agric Food Chem 62:1643–1648
Belščak-Cvitanović A, Durgo K, Bušić A, Franekić J, Komes D (2014) Phytochemical attributes of four conventionally extracted medicinal plants and cytotoxic evaluation of their extracts on human laryngeal carcinoma (HEp2) cells. J Med Food 17:206–217
Nakamura Y, Watanabe S, Miyake N, Kohno H, Osawa T (2003) Dihydrochalcones: evaluation as novel radical scavenging antioxidants. J Agric Food Chem 51:3309–3312
Wright JS, Johnson ER, DiLabio GA (2001) Predicting the activity of phenolic antioxidants: theoretical method, analysis of substituent effects, and application to major families of antioxidants. J Am Chem Soc 123:1173–1183
Leopoldini M, Pitarch IP, Russo N, Toscano M (2004) Structure, conformation, and electronic properties of Apigenin, Luteolin, and Taxifolin antioxidants. a first principle theoretical study. J Phys Chem A 108:92–96
Leopoldini M, Marino T, Russo N, Toscano M (2004) Antioxidant properties of phenolic compounds: H-Atom versus electron transfer mechanism. J Phys Chem A 108:4916–4922
Leopoldini M, Marino T, Russo N, Toscano M (2004) Density functional computations of the energetic and spectroscopic parameters of quercetin and its radicals in the gas-phase and in solvent. Theor Chem Acc 111:210–216
Apak R, Ozyurek M, Guclu K, Capanoglu E (2016) Antioxidant activity/capacity measurement. 2. Hydrogen atom transfer (HAT)-based, mixed-mode (electron transfer (ET)/HAT), and lipid Peroxidation assays. J Agric Food Chem 64:1028–1045
Nenadis N, Sigalas MP (2008) A DFT study on the radical scavenging activity of maritimetin and related aurones. J Phys Chem A 112:12196–12202
Guajardo-Flores D, Serna-Saldivar SO, Gutiérrez-Uribe J A (2013) Evaluation of the antioxidant and antiproliferative activities of extracted saponins and flavonols from germinated black beans (Phaseolus Vulgaris L.) Food Chem 141:1497–1503
Stepanić V, Troselj KG, Lucić B, Marković Z, Amić D (2013) Bond dissociation free energy as a general parameter for flavonoid radical scavenging activity. Food Chem 141:1562–1570
Wright JS, Carpenter DJ, McKay DJ, Ingold KU (1997) Theoretical calculation of substituent effects on the O-H bond strength of phenolic antioxidants related to vitamin E. J Am Chem Soc 119:4245–4252
Brinck T, Lee H -N, Jonsson M (1999) Quantum chemical studies on the thermochemistry of alkyl and peroxyl radicals. J Phys Chem A 103:7094–7104
Gomes JRB, da Silva MAVR (2003) Gas-phase thermodynamic properties of dichlorophenols determined from density functional theory calculations. J Phys Chem A 107:869–874
Vagánek A, Rimarčik J, Lukeš V, Klein E (2012) On the energetics of homolytic and heterolytic O-H bond cleavage in flavonols. Comput Theor Chem 991:192–200
Vagánek A, Rimarčik J, Dropkova K, Lengyel J, Klein E (2014) Reaction enthalpies of O–H bonds splitting-off in flavonoids: the role of non-polar and polar solvent. Comput Theor Chem 1050:31–38
Trouillas P, Marsal P, Siri D, Lazzaroni R, Duroux J -L (2006) A DFT study of the reactivity of OH groups in quercetin and taxifolin antioxidants: the specificity of the 3-OH site. Food Chem 97:679–688
Amić D, Stepanić W, Lučić R, Marković Z, Dmitrić Marković JM (2013) PM6 Study of free radical scavenging mechanisms of flavonoids: why does OH bond dissociation enthalpy effectively represent free radical scavenging activity. J Mol Model 19:2593–2603
Justino GC, Vieira AJSC (2010) Antioxidant mechanisms of Quercetin and Myrcetin in the gas pase and in solution - a comparison and validation of semi-empirical methods. J Mol Model 16:863–876
Antonczak A (2008) Electronic description of four flavonoids revisited by DFT method. J Mol Struct: THEOCHEM 856:38–45
Li M -J, Liu L, Fu Y, Guo Q -X (2007) Accurate bond dissociation enthalpies of popular antioxidants predicted by the ONIOM-g3b3 method. J Mol Struct: THEOCHEM 815:1–9
Lengyel J, Rimarčik J, Vagánek A, Klein E (2013) On the radical scavenging activity of isoflavones: thermodynamics of O-H bond cleavage. Phys Chem Chem Phys 15:10895–10903
Cai W, Chen Y, Xie L, Zhang H, Hou C (2014) Characterization and density functional theory study of the antioxidant activity of quercetins and its sugar-containing analogues. Eur Food Res Technol 238:121–128
Lespade L, Bersion S (2012) Theoretical investigation of the effect of sugar substitution on the antioxidant properties of flavonoids. Free Radic Res 46:346–358
Deepha V, Praveena R, Sivakumar R, Sadasivam K (2014) Experimental and theoretical investigations on the antioxidant activity of isoorientin from Crotalaria globosa. Spectrochim Acta A 121:737–745
Mohajeri A, Asemani SS (2009) Theoretical investigation on antioxidant activity of vitamins and phenolic acids for designing a novel antioxidant. J Mol Struct 930:15–20
de Souza GLC, de Oliveira LMF, Vicari RG, Brown A (2016) A DFT investigation on the structural and antioxidant properties of new isolated interglycosidic O-(1 → 3) linkage flavonols. J Mol Model 22:100–109
Auner BG, Valenta C, Hadgraft J (2003) Influence of phloretin and 6-Ketocholestanol on the skin permeation of Sodium-Fluorescein. J Control Release 89:321–328
Blazso G, Gabor M (1995) Effects of prostaglandin antagonist phloretin derivatives on mouse ear edema induced with different skin irritants. Prostaglandis 50:161–168
Oresajo C (2008) Protective effects of a topical antioxidant mixture containing vitamin C, ferulic acid and phloretin against ultraviolet-induced photodamage in human skin. J Cosmet Dermatol 7:290–297
Rezk BM, Haenen GRMM, van der Vijgh WJF, Bast A (2002) The antioxidant activity of phloretin: the disclosure of a new antioxidant pharmacophore in flavonoids. Biochem Biophys Res Commun 295:9–13
Zhang S, Morris ME (2003) Effects of the flavonoids Biochanin A, Morin, Phloretin, and Silymarin on P-glycoprotein-mediated transport. J Pharmacol Exp Ther 304:1258–1267
Park SY, Kim EJ, Shin HK, Kwon DY, Kim MS, Surh YJ, Park JH (2007) Induction of apoptosis in HT-29 colon cancer cells by phloretin. J Med Food 10:581–586
Luo H, Wang YJ, Liu JQ (2008) Study on the effect of phloretin on inhibiting malignant pheotype of BEL-7402 cells. Zhong Yao Cai 31:1019–1021
Wu CH, Ho YS, Tsai CY, Wang YJ, Tseng H, Wei PL, Lee CH, Liu RS, Lin SY (2009) In vitro and in vivo study of phloretin-induced apoptosis in human liver cancer cells involving inhibition of type II glucose transporter. Int J Cancer 124:2210– 2219
Nithyia T, Udayakumar R (2016) In vitro antioxidant properties of Phloretin—an important phytocompound. J Biosci Med 4:85– 94
Kozlowski D, Trouillas P, Calliste C, Marsal P, Lazzaroni R, Duroux J-L (2007) Density functional theory study of the conformational, electronic, and antioxidant properties of natural chalcones. J Phys Chem A 111:1138–1145
Yan M, Gong J, Shen P, Yang C (2014) The theory investigation for the antioxidant activity of phloretin: a comparation with naringenin. AMM 513:359–362
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Vosko SH, Wilk L, Nusair M (1980) Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can J Phys 58:1200– 1211
Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ (1994) Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J Phys Chem 98:11623–11627
Rassolov V, Pople JA, Ratner M, Redfern PC, Curtiss LA (2001) 6-31G* basis set for third-row atoms. J Comput Chem 22:976– 984
Binkley JS, Pople JA, Hehre WJ (1980) Self-consistent molecular orbital methods. 21. Small split-valence basis sets for first-row elements. J Am Chem Soc 102:939–946
Frisch MJ, Pople JA, Binkley JS (1984) Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J Chem Phys 80:3265–3269
Hehre WJ, Ditchfield R, Pople JA (1972) Self-Consistent Molecular orbital methods. XII. Further extensions of Gaussian-Type basis sets for use in molecular orbital studies of organic molecules. J Chem Phys 56:2257–2261
La Rocca MV, Rutkowski M, Ringeissen S, Gomar J, Frantz M-C, Ngom S, Adamo C (2016) Benchmarking the DFT methodology for assessing antioxidant-related properties: quercetin and edaravone as case studies. J Mol Model 22:250–260
Zhao Y, Truhlar DG (2006) Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. J Chem Theory Comput 2:364–382
Zhao Y, Truhlar DG (2006) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Account 120:215–241
Vydrov OA, Scuseria GE (2006) Assessment of a long range corrected hybrid functional. J Chem Phys 125:234109
Aufmkolk M, Koehrle J, Hesch R-D, Ingbar SH, Cody V (1986) Crystal structure of phlorizin and the iodothyronine deiodinase inhibitory activity of phloretin analogues. Biochem Pharmacol 35:2221–2227
Scalmani G, Frisch MJ (2010) Continuous surface charge polarizable continuum models of solvation. I. General formalism. J Chem Phys 132:114110
Cancès E, Mennucci B, Tomasi J (1997) A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J Chem Phys 107:3032–3041
Mennucci B, Cancès E, Tomasi J (1997) Evaluation of solvent effects in isotropic and anisotropic dielectrics, and in ionic solutions with a unified integral equation method: theoretical bases, computational implementation and numerical applications. J Phys Chem B 101:10506–10517
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian, Inc., Wallingford CT, Gaussian 09, Revision D.01
Brinck T, Haeberlein M, Jonsson M (1997) A computational analysis of substituent effects on the O-H bond dissociation energy in phenols: polar versus radical effects. J Am Chem Soc 119:4239–4244
Kalita D, Kar R, Handique JG (2012) A theoretical study on the antioxidant property of gallic acid and its derivatives. J Theory Comput Chem 11:391–402
Paya M, Goodwin PA, De las Heras B, Hoult JRS (1994) Superoxide scavenging activity in leukocytes and absence of cellular toxicity of a series of coumarins. Biochem Pharmacol 48:445–451
Souza LP, Calegari F, Zarbin AJG, Marcolini-Júnior LH, Bergamini MF (2011) Voltammetric determination of the antioxidant capacity in wine samples using a carbon nanotube modified electrode. J Agric Food Chem 59:7620–7625
Hernández-Herrero JA, Frutos MJ (2014) Colour and antioxidant capacity stability in grape, strawberry and plum peel model juices at different pHs and temperatures. Food Chem 154:199– 204
Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, Windus TL, Dupuis M, Montgomery JA (1993) J Comput Chem 14:1347–1363
Acknowledgments
GLCS thanks Dr. Mariana Pantoja, MD from the Universidade do Estado do Amazonas for useful discussions. This work was partially funded by the Brazilian agency CNPq (Process number: 306266/2016-4). AB thanks the Natural Sciences and Engineering Research Council of Canada for funding (NSERC - Discovery Grant).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mendes, R.A., e Silva, B.L.S., Takeara, R. et al. Probing the antioxidant potential of phloretin and phlorizin through a computational investigation. J Mol Model 24, 101 (2018). https://doi.org/10.1007/s00894-018-3632-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-018-3632-9