
Abstract
Correlated ab initio methods [CASPT2 and R-CCSD(T)] in conjunction with the ANO-RCC basis sets in large contraction were used to calculate potential energy curves (PECs) of the ground and excited electronic states of CsH+ (doublets and quartets) with the inclusion of the scalar relativistic effects and spin-orbit interaction. The ground X2Σ+ state is a rather fragile van der Waals molecular ion. The binding energy of this X2Σ+ state provided by both computational methods is estimated to be 0.02–0.04 eV, and is compared with the reported experimental binding energy (0.51–0.77 eV). This large binding energy can be attributed to the A2Σ+ state, and can thus explain the apparent disagreement between theory and experiment. The spectroscopic constants of all bound states were calculated from the PECs and compared with previous published data for X2Σ+ and A2Σ+ states.

Low-lying Ω states of cesium hydride cation
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Reactions of hydrogen/deuterium and cesium (Cs) are of considerable interest in neutral beam injection (NBI) heating systems, which form an important part of the plasma heating pathway in thermonuclear reactors [1,2,3,4,5,6]. The efficient operation of the NBI source is aided by Cs evaporation and subsequent deposition on a suitable metallic grid. The resulting material should have a low work function to achieve acceptable current densities in the NBI source (102–103 A m−2). For optimal ion source applications and towards a better understanding and control of complex Cs chemistry, it is desirable to accumulate reliable molecular data that can be used in the construction and diagnostics of NBI systems. There is a need for a systematic survey of possible Cs compounds that can be formed from Cs and hydrogen/deuterium. The species considered in this paper can be formed as a result of various types of collisions in the harsh conditions encountered in the NBI heating module under very low pressure (10−6–10−7 mbar) outside the fusion reactor. For instance, electron/CsH, H+/CsH or Cs+/H2 can participate in collisions. In a preceding paper [7], we investigated the manifold of states for neutral CsH and it's a1Σ+→X1Σ+ absorption/emission spectroscopy. Here, we report the results of an ab initio study of the electronic structure of CsH+ in its electronic ground and excited states.
Numerous collisional experiments on CsH+ leading to potential energy curves have been thoroughly reviewed [8]. In the late 1970s and early 1980s, several theoretical papers appeared describing Cs⋯H+ potentials. The common denominator in these papers was the interest in the collision reaction between H+ and Cs, producing the beam of H atoms.
Olson et al. [9] calculated several 2Σ and 2Π states for CsH+ within a simple one-electron model and using a minimal basis set. Their binding energy of >4 eV for CsH+ seems to be overestimated in this model. For the 2Σ+ ground state CsH+, Karo et al. [10] reported a weak attractive interaction as a consequence of charge-induced polarizing effect. Scheidt et al. [11] measured the relative differential cross sections for elastic scattering of H+ by Cs vapor, and derived a binding energy of 0.77 ± 0.05 eV at 10 ± 1 bohr from the Morse model for Cs(6 s)H+. Later, von Szentpály et al. [12] reported the ground-state potential energy curve and spectroscopic constants for 2Σ+ CsH+. Kimura et al. performed electron capture by protons from the ground and excited states of alkali-metal atoms, and also calculated potential energy curves for CsH+ [13]. The results of these early papers on CsH+ are summarized in Table 1.
As can be seen from Table 1, the scatter of both equilibrium distance and binding energy is rather large. There are a few shortcomings associated with previous computational studies. None of them included explicit treatment of correlation/relativistic effects, and the basis sets used were of lower quality, mostly utilizing pseudopotentials. Although the quasi-molecule CsH+ is relatively simple system resembling atom–ion complexes, the determination of its equilibrium structure turned out to be troublesome because the ground state CsH+ exhibits unstable character. According to the majority of previous theoretical studies, CsH+ can be classified as a van der Waals molecule, which certainly puts high demands on the computational protocol when aiming at determining binding energy. For the quantitative description of its bonding, one must resort to high-level calculations, including correlation and relativistic effects. One of our goals was to elucidate the character of X 2Σ+ CsH+: either weak σ-bonding or a van der Waals molecule. Second, we wanted to resolve the apparent discrepancy between theoretical and experimental binding energy, because the high experimental D e (Table 1) implies that it could be attributed to the first excited state rather than to X 2Σ+ CsH+.
This contribution is a follow-up study of neutral CsH with the goal of providing accurate spectroscopic data for the respective cation using reliable model calculations. The material in this paper is organized as follows: we briefly review the Methods used before providing and evaluating the results for the ground and excited states of CsH+ (Results and discussion) and finally, concluding remarks (Conclusion).
Methods
In this study, we used a similar computational strategy as in our precious work [7]. Most calculations were performed with MOLCAS program package [16]. For the ground state, we also adopted the spin-restricted open-shell coupled cluster theory including single, double, and non-iterative triple excitations [17,18,19,20,21], R-CCSD(T) using the MOLPRO program [22]. For the manifold of CsH+ ground and excited states, we used the state-averaged multi-reference complete active space self-consistent field method (SA-CASSCF) followed by multi-state second-order perturbation theory (MS-CASPT2). To avoid underestimation of bond energies, and to suppress the possible intruder states, we used the recommended combination of the IPEA shift (0.25) and small imaginary shift (0.05) [23, 24] in all MS-CASPT2 calculations. For all states, scalar relativistic effects were included with the 2nd order Douglas-Kroll-Hess Hamiltonian (DK) [25, 26]. Spin−orbit coupling was computed within the restricted active space interaction approach employing the SA-CASSCF wave-functions and MS-CASPT2 energies [27]; we denote this as the CASSCF/CASPT2/RASSI scheme. We used the largest contraction of the relativistic ANO-RCC basis sets, hereafter ANO-RCC-L [28]. This contraction of the ANO-RCC basis set can be described as (26s22p15d4f2g)/[12s10p8d4f2g] for Cs and (8s4p3d1f)/[6s4p3d1f] for H. Note that core-valence correlation is included in the design of the ANO-RCC basis, thus it automatically covers the correlation of the 5p shell on cesium.
The complete active space (CAS) was chosen as follows: inactive orbitals 13a and 10b, active orbitals 8a and 6b, giving 9 active electrons in CASSCF step, the same number of electrons was kept active in MS-CASPT2 [29]. This CAS comprises not only the valence space but also a few diffuse orbitals build from atomic Cs[6 s,6p,5d] and H[2 s] shells; these are important for the proper description of higher lying states. In this way, the core-valence correlation (in MS-CASPT2 step) were also included [28]. The active orbitals are (only most dominant AO-contributions are listed): 14a[5 s(Cs)], 15a[5pz(Cs)], 16a[5pz(Cs) + 1 s(H)], 17a[6 s(Cs)-6pz(Cs)-1 s(H)], 18a[6 s(Cs)-2 s(H)], 19a[5dz 2(Cs)-1 s(H)], 20a[5dxy(Cs)], 21a[5dx 2 -y 2(Cs)], 11b[5px(Cs)], 12b[5py(Cs)], 13b[5dxz(Cs)-5dyz(Cs)], 14b[5dxz(Cs) + 5dyz(Cs)], 15[6px(Cs) + 5dxz(Cs)], 15[6py(Cs) + 5dyz(Cs)]. This choice of the active space lead to ~2 × 106 configuration state functions (CSF). The calculations were performed in the reduced symmetry (C 2′ point group). All states were averaged with equal weights. For doublets, we averaged six roots in “A” symmetry, giving four Σ states, one Δ state; and six roots in “B” symmetry, giving three Π states. For quartets, we averaged eight roots in “A” symmetry, giving four Σ states, two Δ states; and ten roots in “B” symmetry, giving five Π states.
The spectroscopic constants: equilibrium distance R e, harmonic frequency ω e, anharmonicity corrections ω e x e, ω e y e, α e, excitation energy T e, and dissociation energy D e were calculated using Dunham analysis [30]. Usually, the 6–8th degree polynomial was used in the fitting procedure. These spectroscopic data were checked against the direct numerical integration that used in the solution of the 1D vibrational Schrödinger equation, and the results from both approaches were numerically consistent.
Results and discussion
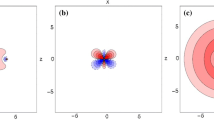
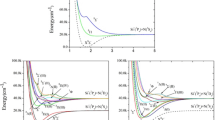
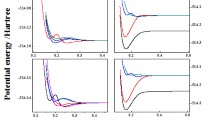
The typical active molecular orbitals (MOs) are displayed in Electronic Supplementary Material, Fig. S1 for the ground state (see the discussion concerning σ-bonding). The contribution to electron density summed up from orbitals along the Cs-H axis is depicted in Fig. S2 (99% of the density is plotted). In Fig. 1 we present MOs for the two lowest excited states of CsH+ (A2Σ+ and B2Π) at equilibrium distance R e = 1.6 Å, which corresponds to the minimum on the potential energy curve. The shapes of active MOs for A2Σ+ and B 2Π are practically identical, although they were obtained from state averaged calculations in two symmetries (for A2Σ+ in “A” and for B 2Π in “B” symmetry, respectively). The active MOs for the distance R e = 3.4 Å (which is the equilibrium distance for the X2Σ+ state) differ only marginally from those depicted in Fig. 1. Spectroscopic constants of the ground and low lying spin-free states of CsH+ are collected in Table 2. Potential energy curves and asymptotes for the Λ doublet states are in Fig. 2, with the zoomed detail of the X2Σ+ potential (in the inset) illustrating the weak bonding in this state. The description of doublet states, including the average occupation of active MOs, and the weights of their leading configurations, is summarized in Table 3. The manifold of quartet states is located high (>13.5 eV) above the manifold of doublet states, in agreement with chemical intuition. All these states are either repulsive or weakly bound; therefore, the pertinent potential energy curves are presented only in Fig. S3. Finally, the low-lying Ω states, i.e., with the spin-orbit interaction included using the RASSI procedure of MOLCAS package [16], are reported in Table 4 and Fig. 3.

Typical molecular orbitals (MOs) in the active space for the A2Σ+/2Π, states of CsH+ at R = 1.6 Å. The numbers correspond to the A2Σ+ natural occupation numbers

Potential energy curves for doublet Λ states of CsH+. Note that the asymptotes of the states F2Σ and G2Π refer to different fragments that are close in energy. Inset Zoom in of detail of the X2Σ+ potential

Detail of the computed potential energy curves of low-lying Ω states
The spectroscopic data for the lowest state indicate that the binding in X2Σ+ CsH+ is dominated by weak ion (Cs+)-induced dipole (on H) interactions. This lowest state can be described as a Cs+⋯H complex with an equilibrium distance ~3.4 Å. The plot of electron density (Fig. S2) has a mid-bond nodal plane, and indicates the absence of regular σ-bonds. When accounting for the zero-point energy contribution, we arrived at the dissociation energy D 0 = 0.02–0.04 eV. Our R-CCSD(T) gave the lower bound of this interval, 0.02 eV, while the MS-CASPT2 calculation led to an upper one of 0.04 eV (cf. inset in Fig. 2). These two approaches are mutually consistent and in good agreement with the dissociation energy reported by von Szentpály et al. [12]. The reason why the experimental data are quite off is because they most probably refer to the first excited state A2Σ+. This state has a potential curve with two minima—deeper “D” at R e = 1.612 Å and more shallow” S” at R e = 4.949 Å, as presented in Fig. 2. In similar cases, one frequently cannot consider spectroscopic parameters of both minima separately, because of tunneling effect through the barrier. However, our A2Σ+ state is of a different kind. The barrier is relatively high, lying more than 0.5 eV above the dissociation limit of the S part. It is also quite broad, as D and S minima are separated by more than 3 Å. We can assume that the tunneling through such a barrier should be very small, and thus both D and S minima appear as independent spectroscopic states, with separate systems of rovibrational levels. Their individual spectroscopic constants (included in Table 1) can serve as a useful tool in experimental identification of these states. We assume that the CsH+ system can exist in spectroscopic states, represented by both minima separately, depending on their means of formation (e.g., ionization, de-excitation, collisional attachment). To verify our assumption of negligible tunneling, we integrated the corresponding radial vibrational Schrödinger equation (rotational quantum number kept at J = 0 level) in the complete region of both minima (1 Å < R < 10 Å). Our technique of direct integration and determination of exact eigenvalues is analogous to that described by Karwowski and Witek [31]. Selected “quasi” S/D vibrational wave functions (i.e., those having significant nonzero values, mainly in the region of S or D minimum) are presented in Fig. 4. To illustrate the order of tunneling, it was useful to switch to the logarithmic scale. For energies corresponding to the pure D/S minimum vibrational levels, we obtained almost identical wave function as in the separated cases, while the probability of tunneling to neighboring minima is by many orders of magnitude smaller. Obviously, the ratio of the wave function amplitudes between “own” and “neighboring” minima (tunneling probability is roughly proportional to the square of this ratio) decreases with increasing vibrational energy, as the separating barrier diminishes. For the lowest vibrational state of D minimum − v D = 0, lying below the bottom of the S part, there is no tunneling at all. For higher v D = 1 state, which is slightly above the S part minimum, we obtained amplitudes ~18 orders of magnitude smaller in the S region, while for v D = 3 level (with the energy about one-half that of the barrier) there are still 10 orders of magnitude difference. For v D = 6 state, which lies less than 0.01 eV below the top of the barrier, and already above the dissociation limit of the S part, we still observe amplitudes in D region that are three orders of magnitude higher than in the outer part, which is no longer bound, and these have a scattering character. The “global” energy eigenvalues are almost indistinguishable from “separate” ones, even for state v D = 6 the difference is below 0.1 cm−1. A completely analogous situation can be found for energy levels corresponding to separate S region vibrational eigenstates.

Vibrational wave functions for selected vibrational states of A 2Σ+ state calculated in complete double minimum potential (see Fig. 2). Logarithmic scale for |Ψv(R)| is employed on the y-axis. States denoted as D are related to the first “Deep” minimum (with R e = 1.612 Å), while those denoted as S are related to the second “Shallow” minimum (with R e = 4.949 Å), with the corresponding vibrational quantum numbers v (for rotational level J = 0). See text for details
When considering the collision processes Cs+ + H in the NBI source, the fragile species, if temporarily formed in the X2Σ+ state, will quickly dissociate back to Cs+ and H. The other possibility to form the cation in the X2Σ+ is ionization from neutral CsH, with the corresponding adiabatic ionization energy (IE) based on our recent results for X1Σ+ CsH [7] and X2Σ+ CsH+, IE(CsH) = 5.49 eV. The possible route to the higher-lying excited states due to electron capture by protons from excited state Cs* atoms was described in [2] by a collision mechanism, but the latter authors did not locate the short-range minima. In our calculations, we revealed the possible existence of states within bonding distances of 1.6–2.2 Å (cf. Fig. 2), some of these states being metastable (A2Σ+, D2Σ+, E2Π). The spectroscopic constants for the three highest states, E2Π, F2Σ+, G2Π, are less accurate estimates due to the avoided crossing between E2Π and G2Π and irregular shape of the F2Σ+ potential around 1.5–1.9 Å. The latter effect is most probably caused by the other higher lying roots, which were not included in the state-averaged group.
Besides the fragile species in the X2Σ+ state, we found a series of higher lying states with typical short equilibrium bond distance in the interval 1.6–2.2 Å. The reason for the contraction of the Cs–H bond is similar as that found for the neutral CsH, but in this case the Cs–H shortening is attributed to the increase in the formal bond order due to single-excitation from anti-bonding MO 16σ with three nodal planes in the X2Σ+ state to either 17σ (A2Σ+) with enhanced electron density in the bond center or to non-bonding 18σ (C2Δ) or to MO 19σ (D2Σ+). Three Σ states, namely A2Σ+, D2Σ+ and F2Σ+, exhibit two avoided crossings, similar to those observed by Kimura et al. [13].
The manifold of quartet states (see Electronic Supplementary Material) is less important, they are too far from the group of doublets, being ~13.5–17.0 eV above the X2Σ+ state; moreover, they are mostly unbound or very weakly bound. We note that, to our knowledge, this is the first exploratory report on CsH+ quartet states.
Table 4 summarizes the spectroscopic constants for the lowest Ω states. We report only a limited number of six Ω states around the corresponding minima. This is because the resolution of the higher lying Ω states was complicated due to the irregular shape of the resulting potential energy curves. The inclusion of spin-orbit (SO) correction to lowest Λ states has led to moderate changes, especially in the shape of B2Π and C2Δ potentials, resulting in Ω potential energy curves as depicted in detail in Fig. 4. The inclusion of spin-orbit interaction modifies the lowest spin-free X2Σ+ state, leading to contraction of equilibrium distance accompanied by slight increase of the bond strength (cf. Table 4, R e and ω e); this is a so-called second-order effect. This effect is even less pronounced in Ω½ (A2Σ+). Spin-orbit splitting of the spin-free B2Π leading to the pair Ω½ (B2Π) and Ω3/2 (B2Π) is small, amounting to 0.038 eV, while the corresponding splitting of the spin-free C 2Δ is larger (0.088 eV). In general, SO coupling in B2Π and C2Δ leads to tightening of the potential, manifested by the increase of harmonic frequency, and, in the case of C2Δ, also to slight shortening of equilibrium bond distance.
Conclusions
We have investigated the potential energy curves of molecular cation CsH+ at the R-CCSD(T)/ANO-RCC and CASPT2/ANO-RCC levels including core-valence correlation and scalar relativistic effects, and demonstrated the consistent performance of both methods for the lowest state. X2Σ+ CsH+ is a typical van der Waals molecule in which an ion-induced dipole mechanism dominates; its binding energy ranges between 0.02–0.04 eV. The measured values: D e of 0.51–0.77 eV and R e 4.76–5.53 Å assigned by experimentalists to the ground state appear to be in good agreement with the D e calculated for the first excited state A2Σ+. Thus, the apparent blatant discrepancy between experiment and theory can be attributed not only to the weakly bound character of the X2Σ+ state, but also to the possible recombination of the colliding particles into the A2Σ+state in scattering experiments. Consequently, all collision experiments could “see” only this state in outer shallow minimum. Measurements at higher angle and/or in higher energy scale could reveal minima with lower R e, but further experimental research is required.
From the point of view of NBI technology, the cation CsH+ will probably play a minor role in plasma heating due to its fragility and the location of the excited states. The manifold of excited states of CsH+ is located quite high above the X2Σ+ state (> 8 eV). The shape of three excited states, A2Σ+, D2Σ+ and F2Σ+ between 2.5 Å and 3.5 Å is the results of avoided crossing. The quartet states of CsH+ are located even higher, above 13.5 eV, and are of minor importance spectroscopically. We have also estimated the ionization energy of X1Σ+ CsH, IE = 5.49 eV from current data for CsH+ and from ref. [7].
References
Gutser R, Wunderlich D, Fantz U (2011) Dynamics of the transport of ionic and atomic cesium in radio frequency-driven ion sources for ITER neutral beam injection. Plasma Phys Control Fusion 53:105014. https://doi.org/10.1088/0741-3335/53/10/105014
Hemsworth R, Decamps H, Graceffa J, Schunke B, Tanaka M, et al. (2009) Status of the ITER heating neutral beam system. Nucl Fusion 49:045006. https://doi.org/10.1088/0029-5515/49/4/045006
Fantz U, Franzen P, Kraus W, Berger M, Christ-Koch S, et al. (2007) Negative ion RF sources for ITER NBI: status of the development and recent achievements. Plasma Phys Contr Fusion 49:B563–B580. https://doi.org/10.1088/0741-3335/49/12b/s53
Heinemann B, Falter H, Fantz U, Franzen P, Fröschle M, et al (2009) Design of the “half-size” ITER neutral beam source for the test facility ELISE. Fusion Eng Design 84:915–922. https://doi.org/10.1016/j.fusengdes.2008.11.076
Kraus W, Fantz U, Franzen P, Froschle M, Heinemann B, et al. (2012) The development of the radio frequency driven negative ion source for neutral beam injectors (invited). Rev Sci Instr 83:02B104–02B105. https://doi.org/10.1063/1.3662957
Fantz U, Franzen P, Wünderlich D (2012) Development of negative hydrogen ion sources for fusion: experiments and modelling. Chem Phys 398:7–16. https://doi.org/10.1016/j.chemphys.2011.05.006
Škoviera J, Neogrády P, Louis F, Pitoňák M, Černušák I (2017) Caesium hydride: MS-CASPT2 potential energy curves and A1Σ+→X1Σ+ absorption/emission spectroscopy. J Chem Phys 146:104304. https://doi.org/10.1063/1.4978065
Stwalley WC, Zemke WT, Yang SC (1991) Spectroscopy and structure of the alkali hydride diatomic molecules and their ions. J Phys Chem Ref Data 20:153–187. https://doi.org/10.1063/1.555906
Olson RE, Shipsey EJ, Browne JC (1976) Theoretical cross sections for H-on-Cs ionic and neutral reactions. Phys Rev A 13:180–195. https://doi.org/10.1103/PhysRevA.13.180
Karo AM, Gardner MA, Hiskes JR (1978) Abinitio MC–SCF ground-state potential energy curves for LiH−, NaH−, and CsH−. J Chem Phys 68:1942–1950. https://doi.org/10.1063/1.435921
Scheidt H, Spiess G, Valance A, Pradel P (1978) Determination of H+ +Cs(6s) potential from differential cross section measurements at energies 13.4-24.2 eV. J Phys B Atomic Mol Phys 11:2665–2685. https://doi.org/10.1088/0022-3700/11/15/015
von Szentpály L, Fuentealba P, Preuss H, Stoll H (1982) Pseudopotential calculations on Rb+ 2, Cs+ 2, RbH+, CsH+ and the mixed alkali dimer ions. Chem Phys Lett 93:555–559. https://doi.org/10.1016/0009-2614(82)83728-7
Kimura M, Olson RE, Pascale J (1982) Molecular treatment of electron capture by protons from the ground and excited states of alkali-metal atoms. Phys Rev A 26:3113–3124. https://doi.org/10.1103/PhysRevA.26.3113
Valance A, Spiess G (1975) Calculation of inelastic cross sections for H++Cs → H(n=2)+Cs+. J Chem Phys 63:1487–1489. https://doi.org/10.1063/1.431512
Sidis V, Kubach C (1978) Theoretical study of the elastic and charge exchange processes in H+ +Cs collision. J Phys B Atomic Mol Phys 11:2687–2703. https://doi.org/10.1088/0022-3700/11/15/016
Aquilante F, De Vico L, Ferre H, Ghigo G, Malmqvist P-Å, et al. (2010) MOLCAS 7: the next generation. J Comput Chem 31:224–247. https://doi.org/10.1002/jcc.21318
Raghavachari K, Trucks GW, Pople JA, Head-Gordon M (1989) A 5th-order perturbation comparison of electron correlation theories. Chem Phys Lett 157:479–483. https://doi.org/10.1016/S0009-2614(89)87395-6
Urban M, Noga J, Cole SJ, Bartlett RJ (1985) Towards a full CCSDT model for electron correlation. J Chem Phys 83:4041–4046. https://doi.org/10.1063/1.449067
Urban M, Černušák I, Kellö V, Noga J (1987) Methods in computational chemistry. In: Wilson SE (ed) Methods in computational chemistry. Electron correlation in atoms and molecules, vol 1. Plenum, New York, pp 117–250
Watts JD, Gauss J, Bartlett RJ (1993) Coupled-cluster methods with noniterative triple excitations for restricted open-Shell Hartree-Fock and other general single determinant reference functions—energies and analytical gradients. J Chem Phys 98:8718–8733. https://doi.org/10.1063/1.464480
Knowles PJ, Hampel C, Werner HJ (1993) Coupled cluster theory for high spin, open shell reference wave functions. J Chem Phys 99:5219–5227. https://doi.org/10.1063/1.465990
Werner HJ, Knowles PJ, Knizia G, Manby FR, Schutz N (2012) MOLPRO, ver. 2012.1, a package of ab initio programs. http://www/molpro.net
Ghigo G, Roos BO, Malmqvist P-Å (2004) A modified definition of the zeroth-order Hamiltonian in multiconfigurational perturbation theory (CASPT2). Chem Phys Lett 396:142–149. https://doi.org/10.1016/j.cplett.2004.08.032
Forsberg N, Malmqvist P-Å (1997) Multiconfiguration perturbation theory with imaginary level shift. Chem Phys Lett 274:196–204. https://doi.org/10.1016/S0009-2614(97)00669-6
Douglas M, Kroll NM (1974) Quantum electrodynamical corrections to the fine structure of helium. Ann Phys (NY) 82:89–155. https://doi.org/10.1016/0003-4916(74)90333-9
Hess BA (1986) Relativistic electronic-structure calculations employing a 2-component no-pair formalism with external-field projection operators. Phys Rev A 33:3742–3748. https://doi.org/10.1103/PhysRevA.33.3742
Roos B, Malmqvist P-A (2004) Relativistic quantum chemistry: the multiconfigurational approach. Phys Chem Chem Phys 6:2919–2927. https://doi.org/10.1039/b401472n
Roos B, Veryazov V, Widmark PO (2004) Relativistic atomic natural orbital type basis sets for the alkaline and alkaline-earth atoms applied to the ground-state potentials for the corresponding dimers. Theor Chem Accounts 111:345–351. https://doi.org/10.1007/s00214-003-0537-0
Walch SP, Bauschlicher Jr CW, Siegbahn PEM, Partridge H (1982) All electron GVB/CI potential curves for the X1Σ+ g state of Cs2. Chem Phys Lett 92:54–58. https://doi.org/10.1016/0009-2614(82)83412-X
Dunham JL (1932) The energy levels of the rotating vibrator. Phys Rev 41:721–731. https://doi.org/10.1103/PhysRev.41.721
Karwowski J, Witek HA (2016) Schrodinger equations with power potentials. Mol Phys 114:932–940. https://doi.org/10.1080/00268976.2015.1115565
Acknowledgments
We thank Slovak Grant Agencies Vedecká grantová agentúra MŠVVaŠ SR a SAV (VEGA) (Grant 1/0092/14) and Agentúra na podporu výskumu a vývoja (APVV) (Project APVV-15-0105). This work was carried out within the framework of the EUROfusion Consortium, and has received funding from the Euratom research and training programme 2014-2018 under Grant Agreement No. 633053. The views and opinions expressed herein do not necessarily reflect those of the European Commission. J.Š. is thankful for the support from the French-Slovak Co-tutelle PhD programme. We thank the Centre de Ressources Informatiques (CRI) of the University of Lille1, the Centre Régional Informatique et d’Applications Numériques de Normandie (CRIANN) and the Research & Development Operational Programme funded by European Regional Development Fund (ERDF) (CGreen-I 26240120001 and CGreen-II 26240120025) for support. J.Š. and F.L. also appreciate support from PIA managed by the French National Research Agency (ANR) under Grant Agreement No. ANR-11-LABX-0005-01 called CaPPA (Chemical and Physical Properties of the Atmosphere) and also supported by the Regional Council “Nord-Pas de Calais” and the “European Funds for Regional Economic Development.”
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper belongs to Topical Collection QUITEL 2016
Electronic supplementary material
ESM 1
(PDF 888 kb)
Rights and permissions
About this article

Cite this article
Škoviera, J., Černušák, I., Louis, F. et al. MS-CASPT2 study of the ground and low lying states of CsH+ . J Mol Model 23, 339 (2017). https://doi.org/10.1007/s00894-017-3503-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-017-3503-9