
Abstract
Three mononuclear coordination complexes of Co(II), Ni(II), and Cu(II) have been synthesized from 2,4-dichloro-6-{(E)-[(5-chloro-2-sulfanylphenyl)imino]methyl}phenol ligand (H 2 L) obtained by simple condensation reaction of 3,5-dichloro-2-hydroxybenzaldehyde and 2-amino-4-chlorobenzenethiol and characterized by elemental analysis, spectral (FT-IR, electronic, and 1H-NMR), molar conductance, thermal, SEM, PXRD, and fluorescence studies. The PXRD analysis and SEM-EDX micrographs show the crystalline nature of complexes. The domain size and the lattice strain of synthesized compounds have been determined according to Williamson–Hall plot. TG of the synthesized complexes illustrates the general decomposition pattern of the complexes. The ligand exhibits an interesting fluorescence property which is suppressed after complex formation. The Co(II) complex adopted a distorted octahedral configuration while Ni(II) and Cu(II) complexes showed square planar geometry around metal center. The geometry optimization, HOMO-LUMO, molecular electrostatic potential map (MEP), and spin density of synthesized compounds have been performed by density functional theory (DFT) method using B3LYP/6-31G and B3LYP/LANL2DZ as basis set.

Three new coordination complexes of Co(II), Ni(II) and Cu(II) with 2,4-dichloro-6-{(E)-[(5-chloro-2 sulfanylphenyl)imino]methyl}phenol Schiff base ligand.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since the start of a new-fangled branch of chemistry established after the Werner’s theory [1]; synthesis and structural characterization of new coordination compounds has always been a challenging task to a chemist. The design and synthesis of a suitable ligand is perhaps the key step in the development of coordination complexes with unique properties and novel reactivity [2–6]. Schiff-base metal complexes derived from salicylaldehyde and its derivatives have been studied extensively in the past and present also due to their facile synthesis, unusual configurations, and structural labiality [7–9].
3d-transition metal ions are found in subtle amounts in living systems but they are directly involved in many life essential biological processes [10]. Numerous Schiff-base metal complexes especially Co(II), Ni(II), and Cu(II) complexes; derived from salicylaldehyde and related derivatives have been studied widely as models for active sites of enzymes [11, 12], as antiviral [13], anti-diabetic [14, 15], anti-inflammatory [16], anticancer drugs [17], and many more. These compounds are successfully utilized as sensor [18], fluorescence materials [19, 20], non-linear optics [21, 22], nano-precursor [23, 24], magnetic materials [25], dye sensitized solar cells [26], OLEDs [27], and have a wide range of catalytic applications such as in polymerization, olefin oxidation, epoxidation, Michael addition reaction, Heck reaction etc. [28–31].
In recent year’s theoretical studies of coordination compounds are also of great interest to explain their structural features and to correlate with properties [32–36]. In the absence of an X-ray crystal structure data, density functional theory (DFT) provides an alternative option for the prediction of fairly accurate three dimensional structures of the compounds [37–39]. Although the field of metal complexes has been explored tremendously, however, the advanced awareness is still sought to target the specific applications. Literature survey reveals that Co(II), Ni(II), and Cu(II) complexes of Schiff base ligands have been extensively studied for their structural diversity and multidimensional applications in materials as well as biological fields[40–43]. It has been noticed that the micro-texture analysis along with thermal and fluorescence studies of Schiff base metal complexes are very rare or omitted; this prompted us to take the present study. This certainly opens a new way to investigate the structural properties of these complexes in future.
In the present paper, the synthesis and spectral characterization of selected Schiff-base ligand H 2 L derived from 3,5- dichloro-2-hydroxylbenzaldehyde and 2-amino-4-chlorobenzenethiol and its mononuclear Co(II), Ni(II), and Cu(II) complexes are reported to investigate the structural properties of Schiff-base ligand H 2 L after complexation. In addition, DFT calculations of synthesized compounds have also been carried out to support the experimental findings.
Experimental
Reagents and instruments
All the reagents were obtained commercially (3,5-dichloro-2-hydroxybenzaldehyde, metal chlorides from Aldrich, India and 2-amino-4-chlorobenzenethiol from TCI Fine Chemicals, India) with analytical grade and used without further purification. Commercial solvents were distilled and then used for the synthesis of Schiff base ligand and its complexes. Microanalysis (C, H, and N) were performed in Thermo Scientific (FLASH 2000) CHN Elemental Analyser (accuracy of 0.3 %.) at Sophisticated Analytical Instrument Facility (SAIF), Panjab University, Chandigarh, India. Molar conductance in DMF (10−3 M) at room temperature was measured using Systronic model-304 digital conductivity meter. The Infra-red spectra were recorded as KBr discs in the range of 400–4000 cm−1 on a Shimadzu FT IR-8400 S instrument. NMR spectra were recorded on a Bruker Ultrashield 500 plus 500 MHz FT-NMR Spectrometer in DMSO-d6 using TMS as the internal reference. The absorption spectra were recorded in DMF using Systronics double beam UV–VIS Spectrophotometer in the range 900–400 nm at room temperature. The fluorescence spectra were obtained in DMF on a Perkin-Elmer LS 55 Fluorescence Spectrometer. Thermal data were collected on a NETZSCH STA instrument under nitrogen atmosphere at the heating rate of 10 °C min−1 from room temperature to 800 °C. X-ray powder diffraction patterns of the samples were recorded at room temperature on a Brucker D-8 Advances X-ray diffractometer using Cu Kα radiation (1.5418 Aο) operating at 35 kV and 25 mA in a step scan mode with a step size of 0.02 (2θ = 5–70°). Surface morphological studies have been performed on scanning electron microscope FEI NOVA Nano SEM 450 with the accelerating voltage of 15 kV at liquid nitrogen atmosphere. Energy dispersive X-ray analysis (EDX) was performed to detect the elements present in the samples that have been coated with gold to make them conductive and then sputtered with carbon black.
Synthesis of 2,4-dichloro-6-{(E)-[(5-chloro-2-sulfanylphenyl)imino]methyl}phenol; ligand (H 2 L)
3,5-dichloro-2-hydroxylbenzaldehyde (1.91 g, 10 mmol) was taken in methanol (25 ml), to which methanolic solution of 2-amino-4-chlorobenzenethiol (1.60 g, 10 mmol) was added drop wise with stirring. Instantly, a yellowish solution was obtained. To facilitate completion of the condensation reaction; stirring was unremitting for 30 min at 60 °C. The progress of reaction was closely examined with the help of TLC. The product thus obtained was filtered off, washed with methanol and ether, recrystallized in methanol and dried in vacuum over fused CaCl2 (Fig. 1).

Synthetic Scheme for Schiff base ligand (H2L) and its metal complexes
Synthesis of the metal complexes [ML2/ ML] {M = Co(II), Ni(II), and Cu (II)}
The metal(II) chlorides (1 mmol), were taken in the minimum volume of methanol possible (~15 ml), and added dropwise to the methanolic solution of H 2 L (1 or 2 mmol) in the presence of aq. KOH solution, the mixture was stirred and refluxed for 1 h. During this period precipitation has started for all of the complexes, and the precipitates were filtered under the hot conditions. The precipitates were washed with methanol and diethyl ether, and dried in vacuum over fused CaCl2.
Theoretical calculations
The entire theoretical calculation of H 2 L and its complexes have been performed at DFT level employing Becke’s three parameter hybrid exchange functional and the Lee–Yang–Parr correlation functional (B3LYP) using the 6-31G and LANL2DZ basis set with the help of Gaussian 03 W and Gauss view 3.0 software [44]. Furthermore, visualization of optimized structures was performed by means of Chemcraft 1.7 program package [45]. The geometry optimization for selected compounds were calculated in a C1 symmetry point group and no symmetry restrains were applied. The frontier molecular orbitals, molecular electrostatic potential map (MEP), spin density are also calculated with B3LYP/6-31G and B3LYP/LANL2DZ basis set.
Results and discussion
The physical and analytical data of synthesized compounds are represented in Table 1. All metal (II) complexes were colored, stable at room temperature and insoluble in most common organic solvents except DMF and DMSO. Analytical data confirms the metal to ligand molar ratio as 1:2 for Co(II) complex while 1:1 for Ni(II) and Cu(II) complexes. The molar conductance measurements of the complexes were recorded in DMF (1 × 10−3 M). The results indicate their non-electrolytic nature.
FT-IR and 1H NMR spectral studies
The FT-IR spectra of the metal complexes have been matched with parental H 2 L to find out the coordination modes involved in chelation (Fig. 2). The characteristic υ (―HC=N―) stretching vibration, found in the H 2 L at 1614.5 cm−1, shifted to lower wave numbers in complexes which indicates the azomethine nitrogen participats in coordination with metal ions (M―N) [46]. The participation of deprotonated phenolic moiety in complexes is verified by the shifting of ν(C―O) stretching band about 15–25 cm−1 from the original; found at 1272.1 cm−1 in the H 2 L after chelation [47, 48]. This may be due to the weakening of ν(C―O) and formation of stronger M―O bond. Coordination with phenolic oxygen and azomethine nitrogen is also confirmed by the appearance of two new bands in the complexes in the range of 522–547 and 445–468 cm−1 which represents the M―O and M―N stretching vibrations, respectively [49]. Involvement of the thiol moiety in chelation is confirmed by the peak shift of υ (C―S) from 1068.6 cm −1 in the H 2 L to the complexes (1078.2–1082.1 cm−1). However, bands corresponding to M―S stretching vibrations appeared below 400 cm −1 and hence cannot be recorded in present studies (Table 2) [50, 51].

FT-IR spectra of H2L and its Co(II), Ni(II), and Cu(II) complexes in the range of 400–4000 cm−1
In Ni(II) and Cu(II) complex two new additional bands appeared about 850–862 cm−1 and 580–590 cm−1 regions due to δr(H2O) rocking and δw(H2O) wagging vibrations respectively from coordinated water molecules [52]. These bands are absent in the H 2 L as well as in Co(II) complex. This is also confirmed by the thermal studies.
The 1H NMR spectrum of the free H 2 L in deuterated DMSO gives two singlets at 8.32 and 9.48 ppm, corresponding to the azomethine (CH = N) and phenolic OH group respectively. The aromatic protons appeared as a set of multiplets in the range 7.82–7.58. The SH signal which appears, in the H 2 L is at 3.37 ppm [53–55].
Electronic spectral studies
Normally the electronic spectra of coordination complexes involving Schiff base ligands does not confirm the molecular geometry around the metal center properly, but they help to support it and tell whether the ligand is chelated or not. The electronic absorption spectra of H 2 L and its complexes were studied in DMF solvent (Fig. 3). The electronic spectrum of the H 2 L showed two absorption bands at 270 and 436 s nm matched up to π–π* and n-π* electronic transitions of the azomethine group. However, these two bands are also observed in the UV–Vis spectra of all the complexes with slight shifts to the higher region in wavelength. This shift may be due to the alteration of molecular environment of H 2 L after the chelation with metal ions [56–58].

Electronic absorption spectra of H2L, Co(II), Ni(II), and Cu(II) complex showing as black, red, blue, and green color respectively in DMF
Powder X-ray diffraction and surface morphological studies (SEM)
Nowadays PXRD are grown to be a powerful tool to get the structural information of materials, where good qualities of single crystals are not yielded. During powder X-ray diffraction analysis; H 2 L and its complexes exhibited sharp peaks showing their crystalline nature as given in Fig. 4. This method is useful to wind up with the structure, size, and strain of the synthesized compounds [59, 60].

PXRD spectra and scanning electron micrographs of (a) H2L, (b) Co(II) complex (c) Ni(II) complex, and (c) Cu(II) complex
The ideal PXRD pattern consists of extremely sharp, symmetric peaks occurred at well set position according to a well-defined unit cell which is obtained by the perfect sample (crystal) and a perfect experimental setup. In general such type of perfections in both the experimental setup and the sample are not possible and typically good powder diffraction patterns display peaks with widths. Ideal crystallinity believes in infinite periodicity, thus no crystal is perfect due to its finite size. This deviation from ideal crystallinity leads to a broadening of the diffraction peaks called crystallite size broadening. Scherrer (1918) is the first one who correlates the peak width to crystalline domain size [61]. Another factor responsible for broadening called lattice strain, or as a more accurate terms we used inhomogeneous strain, arises from displacements of the unit cells about their normal positions. As a result of this the d spacings values are not the same; a compressive stress makes the d spacings smaller whereas a tensile stress makes the d spacings larger [62].
Strain is usually caused by dislocations, surface restructuring, lattice vacancies, interstitials, substitutional etc. and enumerated as ε = Δd/d; where ‘d’ idealized d -spacing and ‘Δ d’ the deviation from d (d + δ d or d -δ d).
The materials at nanoscale level (1–100 nm) usually exhibit some interesting properties, e.g., magnetic, optical, surface reactivity etc. which are not seen when they exist in bulk. By using the peak broadening analysis the information about the microtexture (crystallite size and lattice strain) of a material has been extracted which in turn is useful to understand the size dependent material properties [63].
The crystallite sizes were calculated using the following Scherrer’s equation [61, 64].
where K is a constant and its values varies in the range of 0.62–2.08, D symbolizes the crystallite size (Å), λ is the wavelength of Cu Kα radiation, β is the corrected half width of the diffraction peak and θ is the diffraction angle.
The observed crystallite sizes of H 2 L and its complexes using the above equation are given in Table 3 in nm.
The above method explained the mosaic size only, but does not give any information about the ‘strain’ (ε), since this affects the profile differently in each 2θ value [63, 65]:
where βstrain denotes the variance of the distribution due to ‘strain’ in radians, and ε stands for the ‘strain’.
To overcome the effect of ‘strain’, a well known method called Williamson-Hall can be utilized successfully [66, 67]. This method calculates the size and ‘strain’ separately by two or more orders of a reflection hkl. Thus
where ‘ϒ’ denotes Gaussian component.
If {βsample . cosθ}(1+γ) are plotted versus {4.sinθ}(1+γ), the absolute values of size and strain ‘ε’ can be obtained from the straight regression line (Fig. 5a–d). The micro texture analysis has been carried out with the help of X-Powder software [65].

Williamson-Hall plot using Gaussian method for (a) H2L (b) Co(II), (c) Ni(II), and (d) Cu(II) complex
In profile analysis of H 2 L and its complexes; three methods namely Gaussian, Lorentzian and Pseudo-Voight have been fitted into Williamson-Hall plot to calculate the size and the strain ‘ε’. The results obtained from Lorentzian method find some deviations when matched with the Gaussian and Pseudo-Voight method results. Here results obtained by Gaussian method are discussed as reference out of three methods. The positive strain called tensile strain has been noticed for all the compounds except Cu(II) complex where a negative compressive strain (0.172 %) is introduced. The highest strain (0.263 %) has been found for H 2 L which gradually decreases after chelation in the order of H 2 L > Ni(II) > Co(II). The higher residual strains in the material may be due to the burgeoning of dislocations in the stress fields. The crystallite sizes and strain of all the compounds obtained from Williamson-Hall plot are given in Table 4. The decrease in crystallite size after chelation except Co(II) complex; leads to the possibilities of formation of sub-grain structures within the original grain, hence decreasing the effective crystallite size and broadening the peak profile [63]. It has been observed that the crystallite size and lattice strain (tensile as well as compressive) vary with metal ions and geometry of complexes. Results reveal that the different metal ions and the molecular environment around metal center can cause significant broadening and exhibited noticeable crystallite size and strain in PXRD pattern in a series of complexes derived from a common ligand even adopted a similar synthetic route. To evaluate the size and lattice strain from all three methods Gaussian, Lorentzian, and Pseudo-Voight; the combined Williamson-Hall plots of H 2 L and its complexes are summarized in Fig. S1 and S2 [64, 68].
The SEM analysis has been performed for the surface morphological studies of the H 2 L and its metal complexes and the micrographs obtained are given in Fig. 4 combined with PXRD spectra. The SEM images also support the crystalline nature of all the synthesized compounds moreover each metal complex has a different characteristic surface morphology which is quite different from that of H 2 L. Here H 2 L, Co(II), and Cu(II) complexesare appear as well defined rod shape but of different sizes whereas Ni(II) complex exhibits the oval shape. All the crystals were found to grow up from just a single molecule to several molecules in an aggregate distribution with varying particle sizes [69].
The results obtained by energy dispersive X-ray analysis (EDX) as describes in Fig. S3–S6 have indicated that the residues mainly consist of metal (cobalt, nickel, and copper) and the organic moiety component, e.g., carbon, oxygen, sulfur, and chlorine atoms. These results also favored the microanalytical data of selected compounds [70, 71].
Thermal studies
In a thermogram of complex; degradation starts with the loss of water molecules if present, followed by the ligand moiety in the successive steps. It has been seen that the lattice water molecules were lost firstly in the range of 50–120 °C after that coordinated water molecules were lost at 120–250 °C range in thermal analysis [72, 73]. To evaluate the thermal behavior of metal complexes; the thermogravimetic (TG/DTG) curves for the complexes are represented in Fig. 6 and weight loss at different decomposition stages, temperatures ranges with DTG peaks, assignments, and the final pyrolysis product observed in the present studies are summarized in Table 5.

TG and DTG 3d-plots of Co(II), Ni(II), and Cu(II) complexes showing as orange, white and green color respectively up to 800 °C
In TGA curve of the [CoL2] complex does not show any weight loss up to 380 °C; this indicates the absence of lattice and coordinated water molecules in it. The [CoL2] complex shows two step decomposition process in the range of 370–420 °C and 470–799 °C respectively; the first step is relatively fast and a total of 26.84 % weight loss has been observed which corresponds to the non-chelated part of the ligand moiety. The remaining chelated part of the ligand is decomposed in the second step which is 39.64 % (calc. 38.46 %). The final pyrolysis product, projected as cobalt oxide(III) and some unreacted residue had an observed mass of 33.52 % (calc. 33.34 %). The TG curve of the [Ni(L)(H2O)].H2O complex shows the three steps decomposition process; in the first step 4.52 % (calc.4.23 %) weight loss has been observed in the range of 90–120 °C, which indicates the presence of one lattice water molecule in the complex. Another 4.86 % (calc. 4.23 %) weight loss corresponds to one coordinated water molecule has been observed in the range of 120–170 °C. In the second step of decomposition starting from 310 °C to 440 °C; non-chelated part of the ligand is eliminated which has been found to be 24.82 % (calc. 26.04 %). In the last step starting from 460 °C to the final temperature; 46.84 % (calc. 46.28 %) weight loss corresponds to the remaining part of the ligand has been noticed and the ultimate pyrolysis product is obtained as nickel(II) oxide; of mass 18.96 % (calc.18.03 %). Quite similar results have been recorded for [Cu(L)(H2O)] where decomposition takes place in three steps in the range of 190–270 °C, 290–480 °C, and 500–799 °C respectively and copper (II) oxide; mass of 21.19 % (calc. 19.78 %) seems to be the final product (Fig. 6). The only difference is that no lattice water molecule is presented in [Cu(L)(H2O)] complex. On the basis of TG analysis the increasing order of the stability of complexes as follows [Ni(L)(H2O)].H2O < [Cu(L)(H2O)] < [CoL2]. Thus the Co(II) complex shows excellent thermal stability in all the complexes when subjected to higher temperature hence it is used in such types of applications [74–76].
Fluorescence studies
The fluorescence emission spectra of the H 2 L and synthesized metal complexes have been studied in DMF with excitation wavelength 436 nm at room temperature, given in Fig. 7. Here H 2 L acts as a fluorophore and exhibits a strong fluorescence emission at 481 nm. The interaction of H 2 L with metal ion results either in fluorescence enhancement called chelation-enhanced fluorescence (CHEF) or a decrease in fluorescence called chelation-enhanced quenching (CHEQ). In metal complexes of H 2 L; quenching has been observed with weak fluorescence emission at 483, 486, and 485 nm for Co(II), Ni(II), and Cu(II) complex respectively. The fluorescence emission intensity of H 2 L decrease upon complex formation with metal ion in the order of Co (II) > Cu (II) > Ni(II) which may be due to reduced electron density on ligand H 2 L. Here, it is also noticeable that formation of covalent bonds with metal ion via e − donation from donor atoms to the metal atom changes the emission energy, due to the lowering of the energy gap between molecular orbitals. It seems to disclose that the structural flexibility and the dipole moment of the complex possibly decrease [77–80].

Fluorescence emission spectra of Schiff base ligand, Co(II), Ni(II), and Cu(II) complex showing as black, red, blue, and green color respectively in DMF
Theoretical calculation
DFT calculation has been intensively used to obtain some improved insights into the molecular geometry, electronic structure, and optical behavior of the selected systems in recent years. The gas phase geometries and atomic numbering scheme of H 2 L and its complexes (Fig. 8a–d) were fully optimized with respect to the energy using the B3LYP/6-31G for H 2 L and B3LYP/LANL2DZ basis set for complexes with the help of Gaussian 03 W and Gauss view 3.0 software and the results are summarized in Table 6.

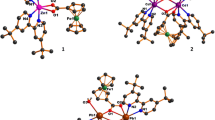
DFT (B3LYP/6-31G and B3LYP/LANL2DZ) optimized structure of (a) Ligand (b) Co(II)-complex (c) Ni(II)-complex (d) Cu(II)-complex showing some selected bond length (A0 ) and bond angles (0)
All selected compounds possess C1 point group. For these compounds, only the ground state geometries have been optimized. Main optimized geometrical parameters, e.g., bond length and bond angles for the H 2 L and complexes are listed in Tables 7 and 8 and detailed information has been summarized in Table S1 and S2.
The DFT calculation of the H 2 L supports the structural assignment from its spectroscopic data. The molecular structure of the ligand and its complexes are shown in Fig. 8a–d, in which selected bond lengths and bond angles are drawn. All the bond lengths and bond angles in phenyl rings are in the common range. The dihedral angle of 33.475° between the two phenyl rings indicates that they are not coplanar. The azomethine group is coplanar with the phenolic ring, as shown by the C2―C3―C9―N11 torsion angle of 0.751°. In ortho-hydroxylated Schiff base ligands, the possibilities of two tautomeric structures as enol-imine and keto-amine forms in the solid state, are generated. In enol-imine tautomer C9―N11 and C2―O21 bonds of the title compound display double bond and single bond character respectively and vice-versa for keto-amine tautomer. In the title compound; the bond lengths between C2―O21 (1.352 A°) and C9―N11 (1.303 A°) and previously reported literature favored the enol-imine form [81]. The former indicates the single-bond character whereas a double-bond character is shown by the later. Furthermore, the H22 atom is located on atom O21, thus H 2 L exists in the enol-imine form in the solid state and the title compound is stabilized by O21―H22∙∙∙∙∙N11 type intramolecular hydrogen bonds [82]. On complexation, C―N, C―O, and C―S bond lengths are altered with respect to that in free H 2 L and are described in Table 1.
The molecular structure of Co(II) complexes shows a distorted octahedral geometry around the Co(II) center as revealed from the calculated bond lengths and bond angles [83].
In Co(II) complex; two ligand moieties are coordinated with metal ion (M:L 1:2) through azomethine nitrogen, phenolic oxygen and through sulfur of thiol moiety in a tridentate fashion. Bond lengths between Co27―N10, Co27―N28, Co27―O24, and Co27―O29 are in a similar range (2.026, 1.954, 1.914, and 1.899 A° respectively), while bond lengths between Co27―S11 and Co27―S30 exhibit a little higher from those values (2.542 and 2.384 A°). The bond angles between N10―Co27―N28, O24―Co27―O29, and S11―Co27―S30 are very close to 180° (178.7°, 175.3°, and 179.3° respectively). This pointed out that N10, O24, N28, and O29 atoms are situated in the corner of the plane and found almost equal distance from the metal center. The remaining two S11 and S30 atoms occupy the up and down positions perpendicular to the plane. The Ni(II) and Cu (II) complexes occupy a square planar geometry with little distortion around the metal center [25, 38, 42]. In Ni(II) complex; three positions of plane are occupied by ligand moiety through azomethine nitrogen, phenolic oxygen and through sulfur of thiol moiety around the metal center while the remaining fourth position is occupied by one molecule of coordinated water. The bond angle between O3―Ni1―O24 is 79.99° which is quite lower as compared to 97.07° for O3―Ni1―N2; 91.22° for N2―Ni1―S4 and 91.72° for S4―Ni1―O24. This may be due to existence of strong intramolecular H-bonding between O3 and H26 (2.119 A°). Bond angles between O3―Ni1―S4 and O24―Ni1―N2 are 177.05° and 171.70° repectively (close to 180°) which suggests the planar structure. Similar observations are seen for Cu(II) complex with slight variations in bond length and bond angles [84, 85].
Frontier molecular orbitals
The HOMO and LUMO are referred to as frontier molecular orbitals (FMOs), without the prior knowledge of FMOs quantum chemistry, optical and electrical properties cannot be executed [86, 87].
The energy gap between HOMO and LUMO energy is the starting point for evaluating a number of global reactivity descriptors. Here the MO energy diagrams are shown the calculated HOMO, HOMO-1, HOMO-2, LUMO, LUMO+1 and LUMO+2 of ligand and its complex and are presented in Fig. 9a and b and Fig. S15 and SS16.

Frontier MO energy diagram showing HOMO, HOMO-1, HOMO-2, LUMO, LUMO+1 and LUMO+2 of (a) Ligand and (b) Ni(II) complex
The HOMO and LUMO energies, the energy gap (ΔE), ionization potential (I), electron affinity (A), absolute electronegativity (χ) chemical hardness (η), softness(S), and electrophilicity index (ω) of the ligand and its complexes are computed at DFT level using B3LYP/6-31G and B3LYP/LANL2DZ basis sets and are listed in Table 9. It has also been noticed that the correlation between the properties and all those calculated parameters are based on purely theoretical assumptions. The HOMO acts as an electron donor, while the LUMO is an electron acceptor. The energy gap (ΔE) makes out the chemical reactivity of compounds [88]. From the results of the calculations as pointed out in Table 9, H 2 L has the largest energy gap (ΔE = 3.72663 e.V.) which comes down after the complex formation and Co (II) complex has the lowest one (ΔE = 1.06561 e.V.) in the series [H 2 L > Cu(II) > Ni(II) > Co(II) ]. Since a higher energy gap entails higher hardness or lower softness [88, 89], the Co(II) complex is referred to as a soft molecule when matched with other compounds.
The chemical potential (I) endows with a global reactivity index arose from charge distribution between two systems having different chemical potential. In the present context all the compounds act as electrophiles thus energy of the system decreases after accepting electrons from the environment and hence their electronic potentials are negative. The electrophilicity index (ω) is another global reactivity descriptor similar to η and I, describes the electron accepting ability of the systems. High value of electrophilicity index makes the molecules a superior electron acceptor. Thus in complexes, electron accepting abilities are arranged in the following order Co(II) > Cu(II) ~ Ni(II).
In the HOMO orbital of H 2 L; the electron density is mainly distributed over thiol group, azomethine group, and associated phenyl rings which is shrunken in the LUMO orbital as shown in Fig. 9a. These FMOs are significantly changed after chelation. In Co(II) complex, HOMO orbital shows that such a type of electron density distribution is shifted over one phenyl ring out of four and very little contribution comes from the metal center which becomes the major part when turned to LUMO as shown in Fig. S15. In Ni(II) and Cu(II) complexes the involvement of metal center is undersized compared to phenyl rings in both HOMO and LUMO orbitals (Fig. 9b and S16). These FMOs compositions are practical to predict which type of transitions will take place in the absorption spectra of the H 2 L and its complex [90].
The spin-density distribution were performed using B3LYP/LANL2DZ basis set for Ni(II) and Cu(II) complex as shown in Fig. 10. In Ni(II) complex the spin density is mainly delocalized into Ni atom and also those atoms which are directly bonded to the metal center. Some part is delocalized on to the carbon atoms of the aromatic ring and two chlorine atoms attached with phenolic moiety of the ligand. This distribution is also satisfied with the HOMO−LUMO shape observed in Ni(II) complex. In Cu(II) complex, the spin density is again distributed over Cu atom and the directly attached atoms with metal center [32] (Fig. 10).

Spin densities in Ni(II) and Cu(II) complexes computed at the B3LYP/LANL2DZ level of DFT theory
Molecular electrostatic potential map (MEP)
In a molecule the electrophilic and nucleophilic sites are easily distinguished with the help of molecular electrostatic potential map (MEP) which are very helpful for unfolding the chemical reactivity type structural properties of a compound. In MEP plots (Fig. S17–S20), blue color represents the upper limit of negative region preferable for electrophilic attack and the red color indicates the utmost positive region favored for nucleophilic attack. Here the negative potentials are seen over the electronegative atoms while the phenyl rings and attached H-atoms have the positive potential region in the structure. In complex the metal centers are preferably electrophilic sites when compared with other atoms [88].
Conclusions
Interaction of H 2 L with metal ions in a tridentate manner resulted in the formation of three new mononuclear complexes. The spectroscopic studies of the resulted Schiff base-metal complexes revealed the structural arrangements. The Cobalt(II) complex exhibits distorted octahedral geometry around metal center whereas nickel(II) and copper(II) complexes occupied square planar geometry with ligand and one water molecule. All the compounds exhibit well defined sharp peaks in PXRD spectra showing their crystalline nature. In micro-texture analysis the crystallite size and strain, which are responsible for line broadening in profile analysis, are determined and are tuned with changing the metal ions and the molecular geometry. Thermal analysis measurements showed a high degree of thermal stability of the complexes in the order of Co (II) > Cu (II) > Ni(II). The H 2 L shows excellent fluorescence properties which are quenched after chelation in the order of H 2 L > Co (II) > Cu (II) > Ni(II). The DFT calculation has also been performed at B3LYP/6-31G and B3LYP/LANL2DZ level for H 2 L and its complexes to support the experimental findings and to evaluate some important parameters viz. bond length, bond angle, HOMO-LUMO, the energy gap (ΔE), molecular electrostatic potential (MEP), chemical hardness (η), softness(S), electrophilicity index(ω), and spin density.
References
Marusak RA , Doan K, Cummings SD (2007) Integrated approach to coordination chemistry an inorganic laboratory guide. Wiley, New York, pp 22–28
Vigato PA, Tamburini S (2004) Coord Chem Rev 248:1717–2128
Vigato PA, Tamburini S (2008) Coord Chem Rev 252:1871–1995
Vigato PA, Peruzzo V, Tamburini S (2012) Coord Chem Rev 256:953–1114
Nelson W, Wallis, Sue C (1974) Cummings Inorg Chem 13:991–994
Cincic D, Kaitner B (2011) Cryst Eng Comm 13:4351–4357
Mishra AP, Jain R (2013) Proc Natl Acad Sci A Phys Sci 83(3):213–223
Uddin AJ, Räisänen MT, Martin N, Markku L, Timo R (2012) Inorg Chim Acta 384:275–280
Bhardwaj VK, Hundal MS, Corbella M, Gomez V, Hundal G (2012) Polyhedron 38:224–234
Rosette M, Roat-Malone (2007) Bioinorganic chemistry a short course, second edn. Wiley, New York, pp 1–28
Dong X, Li Y, Li Z, Cui Y, Zhu H (2012) J Inorg Biochem 108:22–29
Chen W et al (2010) Euro J Med Chem 45:4473–4478
Pelosi G et al (2010) J Med Chem 53:8765–8769
Vanco J et al (2008) J Inorg Biochem 102(4):595–605
Katherine H (2006) Thompson and Chris Orvig. Dalton Trans 761–764. doi:10.1039/b513476e
Shukla S, Mishra AP (2014) Arabian J Chem doi:10.1016/j.arabjc.2014.08.020
Sarah B, Creaven B (2010) Dalton Trans 39:10854–10865
Pandey R et al (2011) Inorg Chem 50:3189–3197
Cozzi PG et al (2003) New J Chem 27:692–697
Strianese M, Milione S, Bertolasi V, Pellecchia C (2013) Inorg Chem 52:11778–11786
Trujillo A et al (2010) Inorg Chem 49:2750–2764
Liu C-G, Guan X-H, Su Z-M (2011) J Phys Chem C 115:6024–6032
Dehno A, Nikookar KM, Fejfarova K, Dusek M (2014) J Mol Str 1071:6–10
Gutiérrez A, Felisa Perpiñán M, Sánchez AE, Carmen Torralba M, Rosario Torres M (2012) Polyhedron 44:165–173
Banerjee S et al (2011) Dalton Trans 40:1652–1661
Nasr-Esfahani M, Zendehdel M, Nia NY, Babadi BJMK (2014) RSC Adv 4:15961–15967
Zhang J et al (2012) J Mater Chem 22:16448–16457
Biswas S et al (2014) RSC Adv 4:34248–34256
Gupta KC, Kumar Sutar A (2008) Coord Chem Rev 252:1420–1450
Cozzi PG (2004) Chem Soc Rev 33:410–421
Su H, Li Z, Huo Q, Guan J, Kan Q (2014) RSC Adv 4:9990–9996
Adhikary J et al (2013) Inorg Chem 52:13442–13452
Das C et al (2014) Inorg Chem 53:11426–11437
Khalaji AD, Chermahini AN, Fejfarova K, Dusek M (2010) Struct Chem 21:153–157
Takaichi J et al (2014) Inorg Chem 53:6159–6169
Lutz OMD et al (2013) J Phys Chem Lett 4:1502–1506
Antunes JA et al (2012) J Mol Str 1013:126–133
Jafarian M et al (2012) J Phys Chem C 116:18518–18532
Upadhyay KK, Kumar A, Upadhyay S, Mishra PC (2008) J Mol Str 873:5–16
Bonhommeau S et al (2012) J Phys Chem C 116:11251–11255
Di Bella S (2001) Chem Soc Rev 30:355–366
Krishnamoorthy P, Sathyadevi P, Muthiah PT, Dharmaraj N (2012) RSC Adv 2:12190–12203
Barone G et al (2013) Coord Chem Rev 257:2848–2862
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T Jr, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, and Pople JA (2004) Gaussian 03, Revision D.01. Gaussian Inc, Wallingford
G A Zhurko; Chemcraft version 1.7(build375) http://www.chemcraftprog.com
Arish D, Sivasankaran M (2010) Nair J Mol Str 983:112–121
Refat MS, El-Sayed MY, Adam AMA (2013) J Mol Str 1038:62–72
Sebastian M et al (2010) Polyhedron 29:3014–3020
Cui Y, Dong X, Li Y, Li Z, Chen W (2013) Euro J Med Chem 58:323–331
Kazuo N (2009) Infrared and Raman spectra of inorganic and coordination compounds part B: applications in coordination, organometallic, and bioinorganic chemistry, sixthth edn. Wiley, Hoboken, pp 210–222
Tyagi P, Chandra S, Saraswat BS (2015) Spectrochim Acta A 134:200–209
Shebl M, Khalil SME, Ahmed SA, Medien HAA (2010) J Mol Str 980:39–50
Singha BK, Prakasha A, Rajourb HK, Bhojakc N, Adhikari D (2010) Spectrochim Acta A 76:376–383
Kavitha P, Saritha M, Laxma Reddy K (2013) Spectrochim Acta A 102:159–168
Soliman AA, Linert W (1999) Thermochim Acta 338:67–75
Ourari A et al (2014) Polyhedron 67:59–64
Sundar A, Prabhu M, Indra Gandhi N, Marappan M, Rajagopal G (2014) Spectrochim Acta A 129:509–518
Klessinger M, Michl J (1995) Excited states and photochemistry of organic molecules, VCH, Weinheim, pp 63–135
Raman N, Mahalakshmi R, Arun T, Packianathan S, Rajkumar R (2014) J Photochem Photobiol B: Biol 138:211–222
Taha A, Farag AAM, Ammarb AH, Ahmed HM (2014) Spectrochim Acta A 130:494–501
Scherrer P (1918) Nachr Ges Wiss Göttinger 2:98–100
Barnes P, Jacques S, Vicker M (2015) Chapter 15: the concept of peak sShape. School of Crystallography, Birkbeck College, University of London. http://pd.chem.ucl.ac.uk/pdnn/relnotes/rel15.htm
Venkateswarlu K, ChandraBose A, Rameshbabu N (2010) Physica B 405:4256–4261. doi:10.1016/j.physb.2010.07.020
Ghosh P, Kar A, Patra A (2010) J Appl Phys 108:113506
Martin JD (2008) X Powder a software package for powder x-ray diffraction analysis user guide ver 2004.04.82. http://www.xpowder.com/download/xpowder.pdf
Balzar D (1993) J Res Natl Inst Stand Technol 98:321–353
Sen Gupta SP, Chatterjee P (2002) PINSA 68(3):267–291
Ghosh P, Patra A (2007) J Phys Chem C 111:7004–7010
Guha A et al (2013) J Mol Str 1042:104–111
Islam SM et al (2011) J Mol Catal A 336:106–114
Abdel-Nasser MA (2014) Alaghaz J Mol Str 1072:103–113
Jain RK, Mishra AP, Gupta P (2012) J Therm Anal Calorim 110(2):529–534
Shukla S, Mishra AP (2012) J Therm Anal Calorim 107:111–117
Alaghaz A-NMA, Ammara YA, Bayoumi HA, Aldhlmani SA (2014) J Mol Str 1074:359–375
Cavalheiro ÉTG et al (2001) Thermochim Acta 370:129–133
Dilek D, Doğan F, Bilici A, Kaya I (2011) Thermochim Acta 518:72–81
Zhou L et al (2012) Anal Chim Acta 735:96–106
Barwiolek M, Szlyk E, Muzioł TM, Lis T (2011) Dalton Trans 40:11012–11022
Singh K, Kumar Y, Puri P, Kumar M, Sharma C (2012) Eur J Med Chem 52:313–321
Patil SA, Naik VH, Kulkarni AD, Badami PS (2010) Spectrochim Acta A 75:347–354
Hameed SA, Alrouby SK, Hilal R (2013) J Mol Model 19:559–569
Tanaka H, Agar AA, Buyukgungor O (2012) Spectrochim Acta A 87:15–24
Gupta SK, Hitchcock PB, Argal GS (2008) Inorg Chim Acta 361:2139–2146
Govindarasu K, Kavitha E (2014) Spectrochim Acta A 133:799–810
Kianfar AH, Ramazani S, Fath RH, Roushani M (2013) Spectrochim Acta A 105:374–382
Ebrahimipour SY, Abaszadeh M, Castro J, Seifi M (2014) Polyhedron 79:138–150
Ghosh M, Weyhermüller T, Wieghardt K (2010) Dalton Trans 39:1996–2007
Yousef TA, El-Gammal OA, Ahmed SF, Abu El-Reash GM (2015) Spectrochim Acta A 135:690–703
Elamurugu Porchelvi E, Muthu S (2015) Spectrochim Acta A 134:453–464
Anbuselvan C, Jayabharathi J, Thanikachalam V, Tamilselvi G (2012) Spectrochim Acta A 97:125–130
Acknowledgments
BSK specially thanks UGC, New Delhi, India, for financial assistance. He is thankful to the Head, Department of Chemistry, Dr. H. S. Gour University, Sagar India, for departmental facilities. He highly acknowledges Mr. Sandeep Tiwari, Department of Physical Science, M.G.C.G. Vishwvidyalaya, Chitrakoot, Satna (M.P.), India and Ms. Anjali Tiwari, Department of Chemistry, Dr. H. S. Gour University, Sagar India for their valuable guidance and motivational support time to time. He is also thankful to Sophisticated Instrumentation Center, Dr. H. S. Gour University, Sagar for making available Powder X-ray Diffraction, thermal analysis and SEM-EDAX facility.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 1922 kb)
Rights and permissions
About this article

Cite this article
Kusmariya, B.S., Mishra, A.P. Theoretical and experimental studies on three new coordination complexes of Co(II), Ni(II), and Cu(II) with 2,4-dichloro-6-{(E)-[(5-chloro-2 sulfanylphenyl)imino]methyl}phenol Schiff base ligand. J Mol Model 21, 278 (2015). https://doi.org/10.1007/s00894-015-2805-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-015-2805-z