
Abstract
Using density-functional calculations, we studied the interaction between interstitial impurities (N, C) and γ-Fe(111) surfaces doped, or not, with Cr, as well as the effect of Cr doping on the dissolution corrosion resistance of the γ-Fe(111) surface. The elementary processes studied afforded microscopic insights into the formation of a Cr-depleted zone, a phenomenon that leads to local corrosion of the stainless steel surface. The aim of this work was to study, at the atomic scale, the effects of N and C on the segregation behavior of Cr and the synergetic effect between co-doped atoms on the resistance to dissolution corrosion of austenitic stainless steel surfaces. The results showed that interstitial impurities prefer to be trapped at near-surface sites, which can impact the segregation behavior of Cr such that it shifts from the surface to the subsurface. Electrode potential calculations and density of states analysis demonstrated that doping with Cr or inserting interstitial impurities into the solid solution can improve the surface corrosion resistance of an fcc Fe substrate, but detrimental effects on the surface corrosion resistance are induced by interactions between Cr and interstitial impurity atoms in co-doped surfaces. The formation of near-surface Cr carbides and nitrides (speculated to be Cr2N and Cr23C6 due to the results obtained for particular co-doped surfaces) was also noted. The results of our theoretical calculations explain some of the experimental results observed at the atomic scale.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Improving both corrosion and mechanical properties is an ongoing goal in the design of stainless steels. Generally, while possessing excellent ductility and fracture toughness, these steels suffer from insufficient yield and ultimate strengths. It is widely accepted that solid-solution strengthening, especially by introducing interstitial atoms such as carbon and nitrogen [1–4], can lead to a favorable combination of strength and toughness. While carbon is the element most commonly alloyed with iron in steels, it is not really appropriate for improving strength and toughness because using carbon lowers the corrosion (particularly the local corrosion) resistance of the steel. Nitrogen has been reported to be an advantageous alloying element in that it improves mechanical properties and resistance to localized corrosion [2, 5, 6]. Thus, nitrogen is commonly added to low-nickel steels to give them high strength, high toughness, and excellent resistance to pitting and crevice corrosion [7, 8]. Nitrogen alloying appeared to enhance the protection afforded by the film on an Fe–20Cr alloy [9]. However, there are also detrimental effects of N on steels.
Austenitic stainless steels (AASs), which are alloys that mainly comprise Fe, Cr, and Ni, are widely used for applications in the biomedical, marine, chemical, power, and oil sectors because of their good localized corrosion resistance. AASs form a dense and protective layer of rhombohedral Cr2O3 over the alloy surface which hampers corrosive attack. When AASs are exposed to severe thermal, chemical, and/or stress conditions, however, local corrosion is also easily induced by the electrochemical potential difference between the matrix and a Cr-depleted zone adjacent to the near-surface precipitation of Cr compounds (generally Cr carbides and nitrides) [10]. It was reported that the gas nitriding of austenitic stainless steels at conventional nitriding temperatures (500–650 °C) resulted in the formation of Cr nitrides, leading to deterioration in the corrosion resistance of the steels, although it also significantly improved their wear properties [3]. Nitrogen interstitial diffusion can induce decomposition in 304L ASS by destabilizing the homogeneous element distribution [11]. H.Y. Ha and H.S. Kwon [12] reported that the near-surface precipitation of lamellar Cr2N has a deleterious effect on pitting corrosion resistance. Although Cr-depletion phenomena at surfaces and grain/interphase boundaries have been observed and investigated in experiments using many different methods [10, 13–18], few theoretical studies of the effects of such phenomena on the corrosion properties of γ-Fe surfaces have been performed [19, 20].
In the work reported in the present paper, in order to elucidate the mechanism for the formation of Cr carbides and nitrides and thus the Cr-depleted zone, as well as the mechanism by which the protective oxide layer is abated or even destroyed (particularly in relation to N and C interstitial impurity alloying), and hence guide the design of high-strength corrosion-resistant austenitic steel, we systematically studied the interactions between interstitial N or C atoms and γ-Fe surfaces doped, or not, with Cr, as well as their effects on surface corrosion properties (i.e., dissolution corrosion) at the atomic scale. We focused on the γ-Fe(111) surface, which contains up to 13.5 % Cr and 0.8–0.9 % N or C. A dense and protective layer of Cr2O3 at the γ-Fe surface is formed when the γ-Fe is doped with Cr atoms. First-principles calculations were performed to study the effects of N and C interstitials on the surface segregation behavior of Cr and on the electrode potential at the surface of the Fe-Cr alloy. Computational methods have been successfully utilized to interpret experimental results, predict properties, and even design materials [21–25]. We hope that the results of the present computational study can help to further our fundamental mechanistic understanding of the effects of Cr, N, and C on austenite surface corrosion at the atomic scale.
Model and method
The present calculations were carried out with the CASTEP code (Cambridge Sequential Total Energy Package) [26], based on density-functional theory (DFT) [27, 28]. The generalized gradient approximation (GGA) via the Perdew–Burke–Ernzerhof (PBE) formula [29] was used for the exchange–correlation potential. The ion–electron interaction was evaluated using ultrasoft pseudopotentials of Vanderbilt type [30]. The cutoff energy for plane-wave expansions was taken as 500 eV. Energy calculations in the first Brillouin zone were conducted using the Monkhorst–Pack scheme [31] with a [6×6×1] k-point mesh grid throughout. Geometry optimization of the structures was performed using the Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm [32], with the following thresholds for converged structures: energy change per atom 1×10−5 eV/atom, maximum force 0.03 eV/Å, maximum stress 0.05 GPa, and maximum displacement 1×10−4 Å. In order to obtain a good compromise between calculation accuracy and efficiency, we used a face-centered-cubic slab consisting of a periodic array of seven Fe layers separated by a vacuum layer of 10 Å which ensured no significant interaction between the slabs, and a (2×2) lateral cell of the fcc Fe(111) surface was employed to model the system of interest (see Fig. 1). Of course, other Fe surfaces such as (110) and (100) are also worthy of corresponding investigations, but we chose the (111) surface because it represents the close-packed plane of γ-Fe, which is the one most likely to occur in actual surfaces. We optimized the coordinates of the top four atomic layers of the slab and the impurities, while the positions of the bottom three layers of Fe atoms were fixed at their positions in the bulk. Full relaxation of atomic positions was carried out until the force on each atom had converged to <0.03 eV/Å. This set of parameters permits a self-consistent field convergence tolerance of up to 1×10−5 eV/atom.

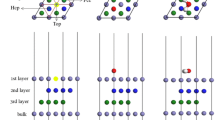
a–b Schematics showing a the fcc Fe(111) surface from the side, b possible interstitial sites (gray) for N or C, and c the most stable surfaces doped with Cr and interstitial N or C (dark blue) as well as the removed Fe atom (sapphire). The green balls represent iron atoms; red balls indicate sites at which Cr substitution has occurred
In order to ensure that the calculations were reliable, the lattice parameters for bulk fcc-Fe were calculated using different functionals (a comparison with the values given in [22] is provided in Table 1). We found that the lattice constant (a) and bulk modulus (B) calculated using GGA-PBE were closest to those reported in [22]. Therefore, GGA-PBE was adopted in subsequent surface relaxation and energy calculations. Although the value we calculated for the lattice parameter (3.446 Å) is very close to some previously reported values (3.45 Å [22] and 3.43 Å [33]), it is significantly different from the experimental value of 3.648 Å [34]. The difference of 0.2 Å is caused by the use of different calculation methods and parameter settings in the two studies. Also note that, given the known paramagnetism of fcc Fe, which is not easy to model within conventional DFT, we chose to model austenite with the nonmagnetic phase of fcc Fe, which leads to further deviation of the theoretical lattice parameter from the experimental value. However, all of the energies we investigated (the segregation energy, adsorption energy, and electrode potential) were relative values obtained by calculating the difference between two or more absolute energy values (all of which are less than the experimentally derived values), thereby decreasing deviations. Furthermore, the surface energy of a clean fcc Fe(111) surface was found to be 3.42 J/m2 using our method, which is in good agreement with other theoretical values (2.83 J/m2 [35] and 3.62 J/m2 [36]), indicating that our calculations are reliable.
In the present calculations, N and C were interstitially implanted into the surface while Cr was doped into the surface via substitution. There are three possible interstitial sites for N and C between neighboring layers: the upright tetrahedral gap (Te1), the reverse tetrahedral gap (Te2), and the octahedral gap (Oct) (see Fig. 1). For all of the Cr-doped γ-Fe(111) surfaces that were modeled by replacing Fe atoms with Cr atoms, substitution occurred according to the sequence marked 1–4 in Fig. 1a as the Cr concentration increased. Although the patterns chosen for substitution and interstitial insertion can affect the results obtained, the patterns we chose were the dominant ones, as the models we used subsequently were the most stable structures.
Results and discussion
Segregation behavior of Cr
Cr-doped surface
It is known that alloying elements may segregate from the bulk to the surface due to the different solubilities of the segregating element at the surface and in the bulk, which can considerably modify the surface properties, leading to positive or negative effects on the corrosion properties of the surface. The segregation of Cr to the surface of the alloy plays a major role in controlling many key properties, including corrosion resistance. For example, it was reported that the surface segregation of chromium can affect the formation of an oxide film on the surface [37]. Thus, we first considered the segregation behavior of Cr in the γ-Fe(111) surface. According to simple thermodynamic considerations, the surface should be enriched by a component with lower surface energy. The empirical surface energies were obtained by extrapolating the results for liquid-metal surface tension to zero temperature, and were found for the (110) surface to be higher for Fe than for Cr: 2.41 and 2.35 J/m2 [38], respectively, suggesting Cr segregation. This is in agreement with the results of our first-principles calculations, which show that the energy of a (111) surface is a little bit higher for Fe than for Cr: 3.42 and 3.20 J/m2, respectively. Besides, the tendency of an alloying atom to segregate to or away from a surface should be dictated by its segregation energy (SE), which can be calculated using the following relation [39]:
where E bulk(nCr) is the total energy of the supercell, which contains n Cr atoms in the bulk, and E ithsurf (nCr) is the total energy of the slab with n Cr atoms moved to the ith surface or subsurface layer. A negative/positive E seg means that Cr does/does not segregate to the surface. Here, it should be noted that we calculated the SE in order to determine the final state or the likely distributions of elements throughout the surface, not to explore the diffusion process in great detail.
The calculated segregation energies are displayed in Fig. 2 as a function of layer depth. For all Cr-doped surfaces, the SE changes nonmonotonically as the surface depth increases; segregation energies in the third layer are much larger than those in the other surface layers, and are positive (they are negative in all the other layers). The SE is lower in a subsurface (second) layer containing one or two Cr atoms than in a surface (first) layer containing one or two Cr atoms, indicating that configurations with Cr atoms in the subsurface are preferred when there are only one or two Cr atoms in the computational cell. However, for higher bulk Cr concentrations, i.e., three and four Cr atoms, the SE is found to be the lowest when the Cr atoms are in the topmost (surface) layer, with an especially high energy barrier observed at the third layer. This result implies the creation of Cr-enriched surfaces, demonstrating that Cr will segregate to the surface as its concentration increases even though a higher temperature is needed for this segregation to occur. This phenomenon was also found to take place in bcc Fe, as also reported previously [39, 40]. The surface segregation of Cr has been confirmed in experiments involving in situ heating at 973 K under an ultrahigh vacuum [37] and the application of conversion electron Mössbauer spectroscopy (CEMS) [41].

Segregation energies of Cr at different layers in a clean γ-Fe(111) surface
Cr-N/C co-doped surface
We also calculated the synergistic effect of the dopants and Fe(111) for an optimized Cr-doped γ-Fe-(111) surface containing interstitial impurities. In order to identify the optimal geometry of a surface doped only with interstitial impurities, we minimized the total energy. Shifts in the total energies of slabs containing a single interstitial atom at different sites (Oct, Te1, and Te2) between every pair of neighboring layers with respect to a slab containing an interstitial atom at the Oct site in the bulk (between the fifth and sixth layers) were calculated by the formula
where E M, surfslab and E Octbulk are the total energies of the slabs with one impurity atom (N or C) at different interstitial sites and at the Oct site in the bulk, respectively. A negative/positive ΔE tot means that the impurity atom does/does not prefer to stay at the corresponding interstitial site compared to its position in the bulk. The calculated total energy shifts are displayed in Fig. 3 as a function of layer depth. In N-doped surfaces, the total energies obtained with different interstitial sites decrease in the order Tet > Oct. In C-doped surfaces, the Tet sites are unstable, so the interstitial C atom will spontaneously migrate from a Tet site to the nearest Oct site. It was found that it is thermodynamically favorable for both N and C to stay at the octahedral site between the second and third layers (i.e., at a near-surface position), as this configuration yields the lowest energy. Based on the resulting models, we investigated the effects of interstitial N and C atoms on the properties of the Cr-doped γ-Fe(111) surface.

a–b Total energy shifts when a N and b C are placed at different interstitial sites
We now consider surfaces that are co-doped with Cr and interstitial atoms (N and C). The impact of a single N or C atom at the Oct site between the second and third layers on Cr segregation was investigated by calculating the segregation energy of Cr via Eq. 1, using a supercell containing an interstitial atom. The calculated segregation energies are displayed in Fig. 4 as a function of layer depth.

a–b Segregation energies of Cr at a γ-Fe(111) surface doped with a N and b C
As we can see, the SE for Cr shows very different trends for the systems with and without an interstitial N or C impurity, indicating that the presence of an interstitial atom affects the surface segregation behavior of Cr. The presence of either an interstitial N or C impurity results in a positive SE value for Cr at the surface and the minimum SE value at the subsurface, regardless of the concentration of Cr. This result obviously suggests that introducing N or C changes the segregation behavior of Cr from surface segregation to near-surface (subsurface) segregation, and this change is not influenced by the Cr concentration. In an N-doped surface, the energy cost of Cr segregation to the subsurface is lower than that in the pure Fe surface case, and even becomes negative, meaning that the introduction of an N interstitial impurity facilitates Cr segregation and can improve the protection provided by the film on the surface by promoting Cr diffusion to the surface. However, Cr segregation is less favored in the C-doped case, as there is a much higher energy cost of segregation to the third layer in this case (0.33 eV) than for a pure Fe surface (0.1 eV), suggesting that the introduction of a C interstitial impurity means that a much higher temperature is required for the segregation of Cr.
Geometric structure and charge properties
The electronic distributions and the bond strengths of the Cr-doped surfaces were modified by the introduction of interstitial N and C impurities, leading to changes in the corrosion properties of the surfaces. Thus, it was necessary to investigate the electronic structures and bonding characteristics of the co-doped surfaces. Therefore, we used Mulliken population analysis and the surface geometries between Cr and the interstitial atom to investigate the effects of N and C on the surface as well as the interaction between them. As shown in Fig. 5, by reviewing the changes in the charges on Cr and C in Cr-C co-doped surfaces (Fig. 5b), we can see that increasing the number of Cr atoms led to more positively charged Cr atoms (Cr co) and more negatively charged C atoms. The Cr–C bond also shortened. All of these phenomena indicate that the interaction between C and Cr strengthens as Cr concentration increases. In N-doped surfaces (Fig. 5a), however, N is most negatively charged and the Cr–N bond distance is shortest for the 2Cr-N co-doped surface, implying that the Cr–N interaction is strongest for this system. These results suggest that the 2Cr-N co-doped surface should be investigated more deeply.

a–b Charges on Cr and the interstitial N and C atoms as well as the bond lengths Cr–N and Cr–C for a Cr-N and b Cr-C co-doped surfaces
Electrode potential shift
Experiments have shown that most steels undergo dissolution corrosion at low oxygen contents (especially <1 × 10−8 wt%) and oxidation corrosion at high oxygen concentrations (>1 x 10−6 wt%). In our work, we also considered the effects N and C interstitials have on the Cr-doped surface in more depth from an electrochemistry perspective, by calculating the electrode potential. Inserting N and C interstitials will cause the surface potential to change, impacting the electrode potential of the system [42]. To study how the presence of interstitial N and C atoms and small nitride and carbide precipitates affects Fe(111) surface dissolution corrosion, the electrode potential shifts ΔU of the Fe–M (M = Cr, N, C) alloy surfaces with respect to the electrode potential of the pure fcc Fe surface for the reaction Fe2+ + 2e− = Fe were calculated using the formula
in which U Fe − M and U Fe are the electrode potentials of the Fe atoms in Fe-M and in the pure fcc Fe(111) surface relative to the standard hydrogen electrode, respectively. The electrode potential can be calculated as follows [43–46]:
where E slab and E slab′ are the total energies of the slab with and without a Fe atom removed from the slab, respectively. μ is the chemical potential of an Fe atom in the slab, which can be evaluated approximately by calculating the total difference in energy between the optimized original slab and the new slab with a vacancy. In our calculations, Fe atoms in the same layer were removed from the slab (see Fig. 1c). As we varied which Fe atom was removed (from the edge midpoint and vertices), the values of U were observed to be rather small, because the distance between the removed Fe atom and the dopants increases. However, the electrode potential shift (∆U) values remained consistent for all co-doped surfaces. Therefore, we only need to guarantee that an Fe atom from a particular layer is removed, as our aim is to simulate the process of Fe dissolution. Namely, the results are independent of the choice of Fe atom to remove. According to this approach, a positive/negative electrode potential shift ∆U indicates a rise/drop in the electrode potential of the Fe-M surface relative to that of the pure Fe surface; we can therefore conclude that adding an alloying element M increases or decreases the ability of the surface to resist electrochemical corrosion.
As shown in Fig. 6, the electrode potential shift (∆U) values are positive for surfaces doped with a single N or C (0.16 V) and for those that are only doped with Cr (0.24 V, 0.15 V, 0.07 V, 0.01 V for 1Cr, 2Cr, 3Cr, 4Cr, respectively), indicating that the presence of N, C, and Cr in solid solution can improve γ-Fe(111) surface dissolution corrosion resistance by suppressing the exit of Fe atoms from the surface, and because doped surfaces are more difficult to electrochemically corrode than a pure Fe surface. Note that the ∆U values of surfaces doped only with Cr decrease with increasing concentration, in contrast to our belief that corrosion resistance improves with increasing concentration of Cr. This result can be ascribed to the models we have used, in which Cr atoms were inserted at the subsurface via substitution, meaning that surface Fe atoms were separated from the substrate by Cr, leading to more Cr atoms in the subsurface and making it easier for Fe atoms to leave the surface. For co-doped surfaces, ∆U values are lower than those for surfaces doped with a single species, but they remain positive except in the cases of 2Cr-N (−0.12 V) and 4Cr-C (−0.45 V). This means that Cr-N or Cr-C co-doping diminishes the beneficial effect of either Cr, N, or C doping on the capacity of the surface to protect Fe atoms from dissolution; indeed, in the cases of 2Cr-N and 4Cr-C, co-doping will actually lead to greater surface erosion. Among the Cr-N co-doped surfaces, the ∆U values of surfaces co-doped with N and 3 or 4 Cr atoms are higher than those of surfaces doped with only 3 or 4 Cr atoms, suggesting that a single N atom can combine with up to two Cr atoms and that these co-doped surfaces may contain Cr nitride precipitates and fewer Cr atoms in solid solution. In the Cr-C co-doped surfaces, a single C atom can combine with up to 4 Cr atoms, resulting in lower ∆U values for all co-doped surfaces and a severe loss of corrosion resistance when C is combined with 4 Cr atoms.

Difference in the electrode potential of an Fe atom in a doped surface from its potential in the pure Fe surface, calculated for various doped surfaces
Based on the above analyses, we can safely conclude that, in solid solution, Cr, N, or C can individually greatly improve the corrosion resistance of the surface. However, combinations of C or N with Cr, especially in 2Cr-N and 4Cr-C co-doped surfaces, degrade the corrosion properties of the surface—an observation that agrees well with results of previous investigations [47–49, 11]. Cr nitrides and carbides may precipitate near the surface, leading to a Cr-depleted zone below the surface, which should be studied in more depth from the perspective of its electronic properties.
Density of states analysis
According to the Fermi–Dirac distribution function, the higher the Fermi energy E f, the lower the electrochemical stability. The Fermi level is located at 0 eV for all of the data. A comparison of the results of density of states (DOS) and partial DOS (PDOS) analysis for the different doped surfaces mentioned above is shown in Fig. 7. The DOS curves for all co-doped surfaces are shown in Fig. 7a, b, while the DOS curves for surfaces doped only with N or C are also plotted for comparison. The DOS analysis suggests that co-doping usually leads to a reduced E f in comparison to single-species doping, except for the 2Cr-N and 4Cr-C co-doped surfaces, for which the E f values are significantly higher than those for single-species-doped surfaces. This result indicates that the introduction of either Cr, N, or C enhances the structural stability of the surface and protects it from being corroded, whereas 2Cr-N or 4Cr-C co-doping destabilizes the structure of the surface and degrades the surface corrosion resistance. We also found that E f is lower for a C-doped surface (24.8 electrons/eV) than an N-doped surface (34.1 electrons/eV), suggesting that doping with C leads to greater corrosion protection than doping with N.

a–d DOS of a Cr-N and b Cr-C co-doped surfaces, and PDOS of c 4Cr-C and d 2Cr-N co-doped surfaces
To further examine the electronic structures of the two special co-doped surfaces 2Cr-N and 4Cr-C, we then focused on the partial density of electronic states (PDOS). This was achieved by decomposing the total density of the single-particle Kohn–Sham eigenstates into contributions from each angular momentum channel of the constituent atoms inside the muffin-tin sphere. In Fig. 7c, d, we can see that the combinations 2Cr-N and 4Cr-C are metallic from the finite DOS at the Fermi level. For both the 2Cr-N and 4Cr-C co-doped surfaces, the DOS can be divided into four main regions: (i) s–p and s–s hybridization regions from N 2s and Cr 3p and from C 2s and Cr 4s for the 2Cr-N and 4Cr-C co-doped surfaces, respectively; (ii) p–d hybridization regions from N/C 2p and Cr 3d, indicating covalent bonding between Cr and interstitial N and C atoms; (iii) d–d bonding states originating from Cr 3d and Fe 3d electrons; (iv) antibonding states above the Fermi level. Hybridization between N or C 2p and Cr 3d electrons plays an important role in the stability of the 2Cr-N combination and that of 4Cr-C. Also, the p–d bond energies are lower for the 4Cr-C co-doped surface (about −6.6 eV) than for the 2Cr-N co-doped surface (about −8.0 eV), implying that the 4Cr-C combination is more stable than 2Cr-N. This suggests that a C atom can more strongly combine with more Cr atoms than an N atom can, leading to substantial weakening of the ability of each dopant species to improve surface corrosion resistance, which can explain why the electrode potential of the 4Cr-C doped surface is lower than that of the 2Cr-N doped surface in Fig. 6. Finally, based on the discussions above, we speculate that the precipitation of Cr nitrides Cr2N and carbides Cr23C6 occurs near to these two special co-doped surfaces, resulting in a Cr-depleted zone and leading to a sudden decrease in pitting corrosion resistance. Also, from the results shown in Fig. 4, Cr2N may precipitate more easily than Cr23C6 near the surface due to the lower energy cost of Cr segregation.
Conclusions
In summary, a plane-wave pseudopotential-based DFT method was employed to study the effects of interstitial N and C impurities on the corrosion properties of γ-Fe(111) surfaces doped, or not, with Cr. Our first-principles calculations showed that, in the absence of interstitial atoms, Cr atoms can become trapped in the topmost layer of the Fe surface as the Cr concentration increases. Generally, when in solid solution, introducing either Cr, N, or C as a dopant can improve the corrosion resistance of the surface. However, the introduction of interstitial N or C atoms along with Cr causes the Cr atoms to tend to segregate towards the subsurface, where N and C prefer to reside. This results in the Cr combining with interstitial atoms, especially in 2Cr-N and 4Cr-C co-doped surfaces, leading to a weakening of the surface corrosion resistance. Based on the special results obtained for 2Cr-N and 4Cr-C co-doped surfaces, we speculate that Cr2N and Cr23C6 form near these surfaces. When they are bound to interstitial N and C atoms, the Cr atoms cannot participate in the formation of a passivating oxide layer, meaning that the corrosion resistance of the co-doped surface inevitably decreases. Analysis of the DOS further demonstrated that interactions between Cr atoms and interstitial N or C atoms weakens the corrosion resistance of surfaces co-doped with them.
References
Tyshchenko AI (2006) Development of stainless austenitic steel alloyed with carbon plus nitrogen. Metallofiz Nov Tekh+ 28:225–233
Bourgin C, Chauveau E, Arnaud A (2006) Effect of a nitrogen addition on mechanical and corrosion properties of 1.4116 martensitic stainless steel. Rev Metall-Paris 103(1):32–36
Christiansen T, Somers MAJ (2005) Low temperature gaseous nitriding and carburising of stainless steel. Surf Eng 21(5–6):445–455. doi:10.1179/174329405x68597
Michal GM, Ernst F, Heuer AH (2006) Carbon paraequilibrium in austenitic stainless steel. Metall Mater Trans A 37A(6):1819–1824. doi:10.1007/s11661-006-0124-9
Stein G, Hucklenbroich I, Feichtinger H (1999) Current and future applications of high nitrogen steels. Mater Sci Forum 318–320:151–160. doi:10.4028/www.scientific.net/MSF.318-320.151
Mudali UK, Khatak HS, Raj B, Uhlemann M (2004) Surface alloying of nitrogen to improve corrosion resistance of steels and stainless steels. Mater Manuf Process 19(1):61–73. doi:10.1081/Amp-120027501
Yang K, Ren YB (2010) Nickel-free austenitic stainless steels for medical applications. Sci Technol Adv Mat 11:014105. doi 10.1088/1468-6996/11/1/014105
Rokanopoulou A, Papadimitriou GD (2011) Production of high nitrogen surfaces on 2205 duplex stainless steel substrate using the PTA technique. Mater Sci Tech-Lond 27(9):1391–1398. doi:10.1179/026708310x12712410311974
Ha H, Jang H, Kwon H, Kim S (2009) Effects of nitrogen on the passivity of Fe-20Cr alloy. Corros Sci 51(1):48–53. doi:10.1016/j.corsci.2008.10.017
Fukunaga T, Kaneko K, Kawano R, Ueda K, Yamada K, Nakada N, Kikuchi M, Barnard JS, Midgley PA (2014) Formation of intergranular M23C6 in sensitized type-347 stainless steel. Isij Int 54(1):148–152. doi:10.2355/isijinternational.54.148
Martinavicius A, Abrasonis G, Scheinost AC, Danoix R, Danoix F, Stinville JC, Talut G, Templier C, Liedke O, Gemming S, Moller W (2012) Nitrogen interstitial diffusion induced decomposition in AISI 304L austenitic stainless steel. Acta Mater 60(10):4065–4076. doi:10.1016/j.actamat.2012.04.014
Ha H, Kwon H (2007) Effects of Cr2N on the pitting corrosion of high nitrogen stainless steels. Electrochim Acta 52(5):2175–2180. doi:10.1016/j.electacta.2006.08.034
Zheng LG, Hu XQ, Kang XH, Li DZ (2013) Precipitation behavior of M23c6 and its effects on ductility and toughness of a noveL Cr-Mn-N austenitic heat resistant steel. Acta Metall Sin 49(9):1081–1088. doi:10.3724/Sp.J.1037.2013.00198
Samal MK, Abhishek A (2011) A new model for the prediction of chromium depletion near grain boundaries and corresponding sensitization in austenitic stainless steels. P I Mech Eng C-J Mec 225(C4):809–815. doi:10.1243/09544062jmes2327
Kaneko K, Fukunaga T, Yamada K, Nakada N, Kikuchi M, Saghi Z, Barnard JS, Midgley PA (2011) Formation of M23C6-type precipitates and chromium-depleted zones in austenite stainless steel. Scripta Mater 65(6):509–512. doi:10.1016/j.scriptamat.2011.06.010
Yu XF, Chen SH, Wang L (2009) Effect of solution treatment conditions on the sensitization of austenitic stainless steel. J Serb Chem Soc 74(11):1293–1302. doi:10.2298/Jsc0911293y
Bennekom PRLA (1995) The involvement of alloyed nitrogen in the corrosion of stainless steels. J South Afric Inst Mining Metall 95(7):337–346
Fu N, Tang XH, Li DY, Parent L, Tian H (2015) In situ investigation of local corrosion at interphase boundary under an electrochemical-atomic force microscope. J Solid State Electr 19(2):337–344. doi:10.1007/s10008-014-2601-1
Tokunaga T, Ohtani H, Agren J (2011) Evaluation of sensitization and self-healing in austenitic stainless steels based on simulations of Cr-depleted zones. Isij Int 51(6):965–968
Yu XF, Chen SH, Wang L (2009) Simulation of recrystallization in cold worked stainless steel and its effect on chromium depletion by cellular automaton. Comput Mater Sci 46(1):66–72. doi:10.1016/j.commatsci.2009.02.008
Riedinger R, Dreyssé H (1985) Electronic structure of interstitial impurities near surfaces. Phys Rev B 31(6):3398–3404. doi:10.1103/PhysRevB.31.3398
Jiang D, Carter E (2003) Carbon dissolution and diffusion in ferrite and austenite from first principles. Phys Rev B 67 (21). doi:10.1103/PhysRevB.67.214103
Yu XF, Chen SH (2009) A simulation of Cr depletion in austenitic stainless steel with cellular automaton. Comput Mater Sci 45(4):899–904. doi:10.1016/j.commatsci.2008.12.012
Turpin T, Dulcy J, Gantois M (2005) Carbon diffusion and phase transformations during gas carburizing of high-alloyed stainless steels: experimental study and theoretical modeling. Metall Mater Trans A 36A(10):2751–2760. doi:10.1007/s11661-005-0271-4
Gavriljuk VG, Shanina BD, Berns H (2008) Ab initio development of a high-strength corrosion-resistant austenitic steel. Acta Mater 56(18):5071–5082. doi:10.1016/j.actamat.2008.06.021
Clark SJ, Segall MD, Pickard CJ, Hasnip PJ, Probert MIJ, Refson K, Payne MC (2005) First principles methods using CASTEP. Z Krist 220(5/6/2005):567–570. doi:10.1524/zkri.220.5.567.65075
Kohn W, Sham LJ (1965) Self-consistent equations including exchange and correlation effects. Phys Rev 140(4A):A1133–A1138
Hohenberg P, Kohn W (1964) Inhomogeneous electron gas. Phys Rev 136(3B):B864–B871
John P, Perdew KB, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:18
Vanderbilt D (1990) Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys Rev B 41(11):7892–7895. doi:10.1103/PhysRevB.41.7892
Monkhorst HJ, Pack JD (1976) Special points for Brillouin-zone integrations. Phys Rev B 13(12):5188–5192
Pfrommer BGMC, Louie SG, Cohen ML (1997) Relaxation of crystals with the quasi-Newton method. J Comp Phys 131:233–240
Häglund J, Fernández Guillermet A, Grimvall G, Körling M (1993) Theory of bonding in transition-metal carbides and nitrides. Phys Rev B 48(16):11685–11691
Basinski ZS, Hume-Rothery W, Sutton AL (1955) The lattice expansion of iron. Proc R Soc Lond Ser A 229(1179):459–467. doi:10.1098/rspa.1955.0102, citeulike-article-id:10762171
Lee S-J, Lee Y-K, Soon A (2012) The austenite/ɛ-martensite interface: a first-principles investigation of the fcc Fe/hcp Fe(0001) system. Appl Surf Sci 258(24):9977–9981. doi:10.1016/j.apsusc.2012.06.059
Jiang Q, Lu HM, Zhao M (2004) Modelling of surface energies of elemental crystals. J Phys Condens Matter 16(4):521–530. doi:10.1088/0953-8984/16/4/001
Suzuki STK, Inoue H, Isshiki M, Waseda Y (1996) Effect of the surface segregation of chromium on oxidation of high-purity Fe-Cr alloys at room temperature. Appl Surf Sci 103:495–502
Tyson WR, Miller WA (1977) Surface free energies of solid metals: estimation from liquid surface tension measurements. Surf Sci 62(1):267–276. doi:10.1016/0039-6028(77)90442-3
Kiejna A, Wachowicz E (2008) Segregation of Cr impurities at bcc iron surfaces: first-principles calculations. Phys Rev B 78:113403. doi:10.1103/PhysRevB.78.113403
Ponomareva AV, Isaev EI, Skorodumova NV, Vekilov YuKh, Abrikosov IA (2007) Surface segregation energy in bcc Fe-rich Fe-Cr alloys. Phys Rev B. doi: 10.1103/PhysRevB.75.245406
Idczak R, Idczak K, Konieczny R (2014) Oxidation and surface segregation of chromium in Fe-Cr alloys studied by Mossbauer and X-ray photoelectron spectroscopy. J Nucl Mater 452(1–3):141–146. doi:10.1016/j.jnucmat.2014.05.003
Zhao W, Wang JD, Lu FB, Chen DR (2009) First principles study of H2O molecule adsorption on Fe(100), Fe(110) and Fe(111) surfaces. Acta Phys Sin-Ch Ed 58(5):3352–3358
Greeley J, Nørskov JK (2007) Electrochemical dissolution of surface alloys in acids: thermodynamic trends from first-principles calculations. Electrochim Acta 52(19):5829–5836. doi:10.1016/j.electacta.2007.02.082
Ma YG, Balbuena PB (2008) Surface properties and dissolution trends of Pt3M alloys in the presence of adsorbates. J Phys Chem C 112(37):14520–14528. doi:10.1021/Jp8046888
Hörmann NG, Jäckle M, Gossenberger F, Roman T, Forster-Tonigold K, Naderian M, Sakong S, Groß A (2015) Some challenges in the first-principles modeling of structures and processes in electrochemical energy storage and transfer. J Power Sources 275:531–538. doi:10.1016/j.jpowsour.2014.10.198
Gossenberger F, Roman T, Groß A (2015) Equilibrium coverage of halides on metal electrodes. Surf Sci 631:17–22. doi:10.1016/j.susc.2014.01.021
Ernst F, Cao Y, Michal GM, Heuer AH (2007) Carbide precipitation in austenitic stainless steel carburized at low temperature. Acta Mater 55(6):1895–1906. doi:10.1016/j.actamat.2006.09.049
Michal G, Ernst F, Kahn H, Cao Y, Oba F, Agarwal N, Heuer A (2006) Carbon supersaturation due to paraequilibrium carburization: stainless steels with greatly improved mechanical properties. Acta Mater 54(6):1597–1606. doi:10.1016/j.actamat.2005.11.029
Zhang ZR, Zhao ZY, Li CZ, Jiang ZH, Li HB (2013) Effects of aging precipitates on the mechanical and corrosion resistance properties of 18Cr-18Mn-2Mo-0.96N super high nitrogen austenitic stainless steel. Appl Mech Mat 395–396:284–288. doi: 10.4028/www.scientific.net/AMM.395-396.284
Acknowledgments
This work was financially supported by the National Science Foundation of China (nos. 51371123, 51304145), the Specialized Research Foundation of the Doctoral Program for Institution of Higher Education (nos. 2013140211003, 20131402120004), the Natural Science Foundation of Shanxi Province (nos. 2014011002–1, 2014021018–1, 2013011010–1, 2013021013–1), and the Opening Fund of the State Key Laboratory of Nonlinear Mechanics.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Han, C., Zhang, C., Liu, X. et al. DFT study of the effects of interstitial impurities on the resistance of Cr-doped γ-Fe(111) surface dissolution corrosion. J Mol Model 21, 206 (2015). https://doi.org/10.1007/s00894-015-2755-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-015-2755-5