
Abstract
We studied microhydrated calcium thiosulfate and its ions at the restricted Hartree–Fock RHF/6-31G* level of theory. A semiempirical molecular dynamics search of progressively more hydrated species provided lowest-energy configurations that were then fully optimized and characterized as energy minima at the RHF/6-31G* level of theory. The first solvation shell of calcium thiosulfate contains 18 water molecules, while the first solvation shell of thiosulfate ion consists of 15 water molecules. QTAIM calculations show hydrogen bonding to sulfur. At 298.15 K, we estimate the total standard enthalpies of hydration for thiosulfate ion and calcium thiosulfate at infinite dilution as −301 kcal mol−1 and −335 kcal mol−1, respectively. The dissociation of hydrated calcium thiosulfate at infinite dilution is predicted to be an endothermic process with an enthalpy of 262 kcal mol−1. Based on some experimental data, the predominant form of calcium thiosulfate in solution is predicted to be the contact ion pair.

Hydrated calcium thiosulfate, showing the first solvation shell
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sulfur oxyanions are of interest because of the industrial and biological applications of their salts. Thiosulfate is used in the paper industry to eliminate chlorine and stop bleaching, and in photography to remove silver bromide from film by complexing silver. It is used in agriculture as a pesticide because it forms sulfur in acid solution, and as a fertilizer when mixed with urea since it produces tetrathionate, which inhibits urea hydrolysis in the soil [1]. Thiosulfate can also be metabolized by bacteria, and has been used as an antidote in cyanide poisoning.
The hydration of ionic species remains a very active area of research [2–4]. According to Liu et al. [5], the solubility of an ionic salt appears to be influenced by the ability of the solvent to produce a solvent-separated ionic pair. Rohman and Mahiuddin [6] experimentally determined a hydration number of 13 for the thiosulfate anion in Na2S2O3. On the other hand, Afanas’ev et al. [7] experimentally found a hydration number of 56 at infinite dilution for Na2S2O3 which, the authors presumed, included water molecules beyond the first solvation shell. Trinapakul et al. [8] used QCMF MD to determine the number of water molecules coordinated by the thiosulfate ion, obtaining a value of 6.8. This value corresponded to a solvation shell radius (from the central sulfur atom) of approximately 3.8 Å, and they observed that coordinating 13 water molecules required a larger solvation shell radius of 4.76 Å.
Knowledge of the structure of the solvation shell allows a greater understanding of the dissolution process as well as the changes in solvation required for a chemical reaction, but this information is not always available. For example, Nelson et al. [9] reported a substantial influence of hydration effects on the complexation of thiosulfate by cryptates, but had to resort to qualitative arguments because no hydration energies for thiosulfate have been reported. Steudel and Steudel [10] reported hydration energies for thiosulfuric acid and its monoanion with up to three water molecules at the G3X(MP2)//B3LYP/6-31G(2df,p) level of theory. They found a hydration energy of −51 kJ mol−1 per water molecule. We did not find any systematic studies of the progressive hydration of thiosulfate performed at an ab initio level using a solvent model of explicit water molecules up to or beyond the solvation shell, and thought that such a study was needed to provide a reference point that allows a more precise determination of the solvation shell in thiosulfate.
We previously reported [11] a systematic study of the microsolvation of calcium oxalate and its ions that followed a very similar procedure. Here, we continue with our studies of the solvation of calcium salts by looking at a completely inorganic solute with a partially hydrophobic region and a high charge density on the oxygens (in comparison with the oxalate ion, which has two regions of low charge density, one at each end). It is interesting to note that thiosulfate is fairly soluble in water, which is counterintuitive given the presence of a partially hydrophobic region in the molecule.
Methods
Software employed
Molecular structures were built using Ghemical [12] or Avogadro [13]. Semiempirical molecular dynamics and preoptimizations employed the PM6 [14] Hamiltonian implemented in MOPAC2012 [15]. Optimizations at the RHF/6-31G* level of theory used GAMESS-US [16] and ORCA v.2.9 [17]. Initial configurations for the Monte Carlo-like procedure were generated with PACKMOL [18]. Atom–atom distances were determined using Jmol [19]. Images were produced using Jmol and ray-traced by POV-Ray [20]. Cropping and color-to-grayscale transformation of the ray-traced images used GIMP v.2.6.7 [21].
The procedures used for cluster structure optimization and determining the hydration shell structure have been previously described in another publication [11], so we only provide a summary here. According to Kelly [22], one of the most challenging aspects of solvation energy calculation is to account for the effect of the solvent in a physical way. Truhlar [22] and Tang et al. [23]—among others—have resorted to a continuum solvent model supplemented with explicit solvent molecules. We have chosen a different route to achieve this physical description of the solvent effect, comprising a configurational search of explicit solvent molecules around the solute by means of semiempirical molecular dynamics.
Even though the HF/6-31G* level of theory may seem low for these systems, we have previously shown [11] that the average unsigned error resulting from the application of HF/6-31G* vs. MP2/def2-SVP to calculate the geometric parameters of these systems is about 2.6 %, so we continuedto use the HF/6-31G* level of theory due to the size of the systems we studied.
Cluster structure optimization
We employed two general procedures, one based on molecular dynamics and the other on a Monte-Carlo-like configurational search. Both methods involved building the molecular structure using Ghemical and performing geometric optimization of that structure using the semiempirical Hamiltonian PM6. The methods then diverged. The molecular dynamics search consisted of (a) taking the optimum semiempirical structure obtained, loading it back into Ghemical and adding one water molecule, (b) finding the global minimum of the new structure via semiempirical molecular dynamics, and (c) selecting the lowest-energy structure and repeating the procedure from step (a) up to the total number of water molecules considered for each species. The Monte-Carlo-like procedure involved using PACKMOL to generate 50–100 initial configurations of water molecules around the calcium thiosulfate (a different random seed was used in each case), and the resulting configurations were then optimized at the RHF/6-31G* level of theory and their vibrational frequencies calculated.
Determination of bond critical points by QTAIM
All the bond critical points were determined by Multiwfn [24]. Critical points were found by searching by nuclear positions, by midpoints, by triangles, and by tetrahedra until no new critical points were found.
Determination of hydration shell structure
For the global minimum of the most hydrated structure obtained at the ab initio level, we determined the radial distribution function (RDF) of the water molecules using the oxygen (Ow) as a reference point for each water molecule and the geometric centroid of the molecule as a reference point for the solvation shell around said molecule. For each RDF, we computed the running coordination number.
To determine the presence of a hydrogen bond, we adopted the geometric criteria from Xenides et al. [25], and—as stated by Desiraju [26]—the presence of a (3,−1) bond critical point by QTAIM [27] calculations.
Energy calculations and corrections
All the Hartree–Fock energies were computed at 298.15 K and corrected for zero-point energy (ZPE). Enthalpy values were calculated by taking the thermal corrections reported in the thermochemical analysis section and adding them to the ZPE-corrected total energy [28, 29].
Total (Δhyd H°) and differential (successive) (Δdiff H°) standard hydration enthalpies were calculated using Eqs. 1 and 2, respectively, for the processes shown in Scheme 1:

Reactions used for the calculation of total standard hydration enthalpy and differential standard hydration enthalpy
The step-wise binding electronic energy (Δstep E) was calculated using
Upon dissociation, the solvating water molecules in calcium thiosulfate are split among the ions according to Scheme 2, where n = 1–19.

Distribution of solvation water molecules between dissociated ions
The conditions used to choose the value of m are based on the work of Woon and Dunning [30], who showed that water preferentially solvates the cation rather than the anion when adding water molecules, so the first six water molecules were used to solvate the Ca(II). Dissociation enthalpies (Δdiss H°) were calculated by
Results
Structures of solvated species and solvation shells
From Table 1, we can see that, as the number of water molecules increases, the S–S bond increases to 1.99 Å, a value that agrees nicely with that calculated by Trinapakul et al. (1.981 Å) [8]. The S–O bond lengths develop some asymmetry as solvation progresses, but when 19 water molecules are bound, the S–O bond lengths are very close to being the same length.
Now, looking at Table 1, the situation is different for calcium–oxygen distances, as these distances do not return to being equal in length upon increased solvation, and they end up being asymmetric. This is probably due to the effect of H-bonding on the thiosulfate oxygens, because all three oxygen atoms in thiosulfate act as donors in different numbers of hydrogen bonds (see Fig. 1), so their abilities to compensate for the positive charge on Ca(II) are hindered to differing degrees by the H-bonding. As a whole, the Ca(II) gets farther away from the thiosulfate—as shown by the Ca–Scentral distance values—as solvation increases, but it does not form solvent-separated ion pairs, even with 19 water molecules.

Hydrogen bond network for CaS2O3 with 19 water molecules (left), and for S2O3 2− with 16 water molecules (right). The aqua-colored spheres are (3,−1) bond critical points corresponding to H-bonds. The dark orange spheres are (3,−1) bond critical points corresponding to interactions of water hydrogens with sulfur. All the other (3,−1) bond critical points have been hidden for clarity
From Fig. 2 we observe that all the ion pairs are contact ion pairs. We can also see that water is located away from the external sulfur atom and closer to the calcium.

Optimum structures for CaS2O3(H2O) n , n = 0–19, calculated at the RHF/6-31G* level of theory
The radial distribution function (see Fig. 3) shows that the first solvation shell in CaS2O3(H2O)19 extends to approximately 6.4 Å and contains 18 water molecules. From Table 2, we can see that the hydration energy for 18 water molecules is −240.41 kcal mol−1. This yields −13.3 kcal mol−1 per water molecule, or approx. −56 kJ mol−1. This value is in the upper limit of the range reported by Steudel [10] (−51 ± 5 kJ mol−1 per water molecule), who calculated it at a higher level of theory.

Radial distribution function M–Ow for the cluster CaS2O3(H2O)19, calculated for the optimum structure obtained at the RHF/6-31G* level of theory (M is the geometric centroid of CaS2O3 and Ow is the oxygen atom in the water molecules)
How do hydrogen bonds help stabilize the solvation shells? The oxygens in thiosulfate are H-bond acceptors and direct the formation of the solvation shell: the first solvating water is H-bonded to an oxygen in thiosulfate for both calcium thiosulfate and the anion (see Fig. 4). It is interesting to note the presence of a (3,−1) bond critical point between water and the external sulfur in the thiosulfate ion. Hydrogen bonds to sulfur will be discussed later on.

First water of solvation for CaS2O3 (left) and S2O3 (right). The aqua spheres are (3,−1) bond critical points corresponding to H-bonds. The dark orange sphere is a (3,−1) bond critical point corresponding to the interaction of a water hydrogen with sulfur. All the other (3,−1) bond critical points have been hidden for clarity
Hydrogen bonding helps in the stabilization of the solvation shells, mostly by forming a network of water–water hydrogen bonds. The oxygen atoms in the thiosulfate participate in a few H-bonds, but the number of thiosulfate–water H-bonds is between one-quarter and one-half of the number of water–water H-bonds, as we can see from Tables 1 and 3.
For calcium thiosulfate, water–water hydrogen bonds start appearing with four molecules of water, while for the anion this happens with three molecules of water. As the number of water molecules increases, water–water H-bonds appear to contribute more to the stabilization of the solvation shell, given that with 10 water molecules, there are already 10 H-bonds, and when 19 water molecules are present, 24 water–water H-bonds have formed. For the anion, when n = 16, 19 water–water H-bonds have formed (see Fig. 1).
Each hydrogen bond determined by geometric criteria has a corresponding bond critical point. This is remarkable in light of the report by Lane [31] which shows that bond critical points do not always appear in cases where other evidence clearly points to the presence of a hydrogen bond.
From Figs. 2 and 5 we see that—even when fully solvated—the surroundings of the external sulfur atom are devoid of water molecules, so the external sulfur seems to define a hydrophobic site. This is consistent with the work of Trinapakul [8], who found a negligible influence of the external sulfur on the hydration shell. However, a more detailed analysis reveals some hydrogen bonding of the water molecules to the external sulfur atom. According to several reported crystal structures of diverse thiosulfate salts, hydrogen bonds between water and the external sulfur atom range from 3.24 Å [32] to 3.298 Å [33] and up to 3.359 Å [34] in length. By looking at the bond critical points, we find an additional four hydrogen bonds, now between water and the external sulfur. In all four cases, the hydrogen bond to the external sulfur is almost perpendicular to the S–S bond (see Fig. 6).

Optimum structures for S2O3 2−(H2O) n , n = 1−16, calculated at the RHF/6-31G* level of theory

Example of the arrangement of water–sulfur hydrogen bonds, showing the plane perpendicular to the S–S bond and the external sulfur atom highlighted as a sphere. The dark orange spheres are (3,−1) bond critical points corresponding to interactions of water hydrogens with sulfur. All the other (3,−1) bond critical points have been hidden for clarity
According to Platts et al. [35], who studied hydrogen bonding to sulfur on hydrogen sulfide, this more perpendicular approach is due to the participation of a large charge-quadrupole component in H···S hydrogen bond, while more traditional hydrogen bonding to oxygen is dominated by charge–charge interactions. This helps explain the origin of the hydrophobic site found by Trinapakul et al. [8]. There is little incentive to form a hydrogen bond linear to the S–S bond because the terms that drive the formation of the hydrogen bond on sulfur (monopole–dipole and the monopole–quadrupole) prefer an orientation of 90° off the S–S bond axis. This leaves a relatively large volume around the external sulfur empty, as there is no driving force to form hydrogen bonds there.
From Fig. 7, we observe that the first hydration shell in S2O3 2−(H2O)16 extends to approximately 5.6 Å from the geometric centroid of the thiosulfate ion and contains 15 water molecules. This is somewhat larger than the value of 13 estimated by Rohman and Mahiuddin [6], but due to the difficulty involved in experimental determination, we think the agreement is acceptable.

Radial distribution function M–Ow for the cluster S2O3 2−(H2O)16, calculated for the optimum structure obtained at the RHF/6-31G* level of theory (M is the geometric centroid of S2O3 2− and Ow is the oxygen atom of the water molecules)
Hydration energies at infinite dilution
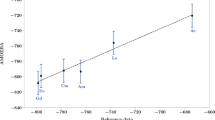
Table 2 and Fig. 8 show the hydration enthalpies for S2O3 2− and CaS2O3 for the processes
and

Total standard hydration enthalpy (∆hyd H°) at 298.15 K, in kcal mol−1, for the hydration processes Ca2+ + nH2O → Ca2+(H2O) n , n = 0–14, S2O3 2−(H2O) n−1 + H2O → S2O3 2−(H2O) n , n = 1−16, and CaS2O3 + nH2O → CaS2O3(H2O) n , n = 1–19, calculated at the RHF/6-31G* level of theory
The data for the hydration of Ca(II) were taken from our previous work [11] for ease of comparison. We can see that as n increases, the average hydration enthalpy per water is reduced. To estimate hydration enthalpies at infinite dilution, we fit the hydration enthalpies to equations of the form
where n is the number of water molecules while a, b, and c are fitting parameters.
As previously reported [11], the n + 3 factor resulted from minimization of the standard error in c. The parameters resulting from the fit are gathered in Table 4.
By extrapolating to the limit of infinite dilution (n → ∞), using Eq. 5, we can interpret c as equal to the enthalpy change of hydration at infinite dilution (∆hyd H°∞). From this, we can estimate the hydration enthalpy of CaS2O3 at infinite dilution as −335 kcal mol−1, and that for S2O3 2− as −301 kcal mol−1.
Energies of dissociation
Figure 9 depicts the trends in the dissociation enthalpies of CaS2O3(H2O) n clusters with increasing n, based on the data in Table S1 (see the Electronic supplementary material, ESM). The energetic cost of separating the ions of CaS2O3 in the gas phase is 468.33 kcal mol−1. When 6 molecules of water are added, dissociation costs 361.80 kcal mol−1. Upon complete hydration, i.e., each ion has a full solvation shell, dissociation costs 306.55 kcal mol−1. By fitting the data in Table 5 to Eq. 5 (a = 1067.61, b = −1344.41, c = 261.92, R 2 = 0.9986, standard error in c = 1.5), we find the predicted enthalpy of dissociation at the limit of infinite dilution to be close to 262 kcal mol−1. So, even at the limit of infinite dilution, the enthalpy of dissociation is positive. Given that calcium thiosulfate has a very high solubility in water (for CaS2O3·6H2O, 92 g/100 mL of water at 25 °C [36]), it seems that either the dissociation involves a very favorable entropic change, or the solvated undissociated ion pair is the predominant form in solution. Experimental data point to the second option as the correct one, as the association constant of the solvated ion pair—according to Gimblett and Monk [37]—is approximately 80.

Dissociation enthalpies (in kcal mol−1) for the hydration process CaS2O3(H2O) n → Ca2+(H2O) m + S2O3 2−(H2O) n−m clusters, n = 0–19, 298.15 K. The vertical arrows include the enthalpies of solvation and formation of the water molecules (−47666.30 kcal mol−1) required to form the complex
Conclusions
According to our calculations, which included a configurational search for the global minimum, the first solvation shell in CaS2O3 contains 18 water molecules, while that of S2O3 2− contains 15 water molecules. H-bonding in the solvation shells of both thiosulfate ion and calcium thiosulfate is preferentially directed towards the negatively charged oxygen atoms. Due to its partially neutralized negative charge, the thiosulfate oxygens in calcium thiosulfate form fewer hydrogen bonds with water than those on the thiosulfate ion. Hydrogen bonding to the external sulfur in thiosulfate seems restricted to the plane perpendicular to the S–S bond, because the charge components involved prefer this orientation, so most of the external sulfur surface is not amenable to hydrogen bonding. Water–water H-bonding starts with n = 4 for calcium thiosulfate and n = 3 for the thiosulfate ion.
The total standard solvation enthalpies at the HF/6-31G* level approach asymptotic limits of −301 kcal mol−1 for the thiosulfate anion and −335 kcal mol−1 for calcium thiosulfate. These are the first estimates of solvation enthalpies for these species. The estimated enthalpy of dissociation of solvated calcium thiosulfate (CaS2O3(H2O) n ) into solvated ions is always endothermic for n = 1–19, with an asymptotic limit of 262 kcal mol−1 at infinite dilution.
References
Sullivan DM, Havlin JL (1992) Thiosulfate inhibition of urea hydrolysis in soils-tetrathionate as a urease inhibitor. Soil Sci Soc Am J 56:957–960. doi:10.2136/sssaj1992.03615995005600030045x
Ohtaki H, Radnai T (1993) Structure and dynamics of hydrated ions. Chem Rev 93:1157–1204. doi:10.1021/cr00019a014
Robertson WH, Johnson MA (2003) Molecular aspects of halide ion hydration: the cluster approach. Annu Rev Phys Chem 54:173–213. doi:10.1146/annurev.physchem.54.011002.103801
Marcus Y (2009) Effect of ions on the structure of water: structure making and breaking. Chem Rev 109:1346–1370. doi:10.1021/cr8003
Liu C-W, Wang F, Yang L, Li X-Z, Zheng W-J, Gao YQ (2014) Stable salt–water cluster structures reflect the delicate competition between ion–water and water–water interactions. J Phys Chem B 118:743–751. doi:10.1021/jp408439j
Rohman N, Mahiuddin S (1997) Concentration and temperature dependence of ultrasonic velocity and isentropic compressibility in aqueous sodium nitrate and sodium thiosulfate solutions. J Chem Soc Faraday Trans 93:2053–2056. doi:10.1039/a700022g
Afanas’ev VN, Tyunina EY (2004) Structural features of ion hydration in sodium nitrate and thiosulfate. Russ J Gen Chem 74:673–678
Trinapakul M, Kritayakornupong C, Tongraar A, Vchirawongkwin V (2013) Active site of the solvated thiosulfate ion characterized by hydration structures and dynamics. Dalton Trans 42:10807–10817. doi:10.1039/c3dt50329a
Nelson J, Nieuwenhuyzen M, Pál I, Town RM (2004) Dinegative tetrahedral oxoanion complexation; structural and solution phase observations. Dalton Trans 15:2303–2308. doi:10.1039/b401200c
Steudel R, Steudel Y (2009) Microsolvation of thiosulfuric acid and its tautomeric anions [HSSO3]− and [SSO2(OH)]− studied by B3LYP-PCM and G3X(MP2) calculations. J Phys Chem A 113:9920–9933. doi:10.1021/jp905264c
Rosas-García VM, Sáenz-Tavera IC, Rodríguez-Herrera VJ, Garza-Campos BR (2013) Microsolvation and hydration enthalpies of CaC2O4(H2O) n (n = 0–16) and C2O4 2−(H2O) n (n = 0–14): an ab initio study. J Mol Model 19:1459–1471. doi:10.1007/s00894-012-1707-6
Hassinen T, Peräkylä M (2001) New energy terms for reduced protein models implemented in an off-lattice force field. J Comput Chem 22:1229–1242. doi:10.1002/jcc.1080
Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR (2012) Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform 4:17. doi:10.1186/1758-2946-4-17
Stewart JJP (2007) Optimization of parameters for semiempirical methods. V: Modification of NDDO approximations and application to 70 elements. J Mol Model 13:1173–1213. doi:10.1007/s00894-007-0233-4
Stewart JJP (2012) MOPAC2012. Stewart Computational Chemistry, Colorado Springs. http://OpenMOPAC.net, last date of access 15th Jan 2015
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, Windus TL, Dupuis M, Montgomery JA Jr (1993) General atomic and molecular electronic structure system. J Comput Chem 14:1347–1363. doi:10.1002/jcc.540141112
Neese F (2012) The ORCA program system. Wiley Interdiscip Rev Comput Mol Sci 2:73–78. doi:10.1002/wcms.81
Martínez L, Andrade R, Birgin EG, Martínez JM (2009) PACKMOL: a package for building initial configurations for molecular dynamics simulations. J Comput Chem 30:2157–2164. doi:10.1002/jcc.21224
Jmol Development Team (2012) Jmol: a 3D molecular visualizer. http://www.jmol.org, last accessed 15 Jan 2015
Persistence of Vision Pty. Ltd. (2004) Persistence of Vision raytracer (version 3.6). http://www.povray.org/download/, last accessed 15 Jan 2015
GIMP Development Team (2009) GNU Image Manipulation Program (GIMP) v.2.6.7. http://www.gimp.org, last date of access 15th Jan 2015
Kelly CP, Cramer CJ, Truhlar DG (2006) Adding explicit solvent molecules to continuum solvent calculations for the calculation of aqueous acid dissociation constants. J Phys Chem A 110:2493–2499. doi:10.1021/jp055336f
Tang E, Di Tommaso D, de Leeuw NH (2010) Accuracy of the microsolvation–continuum approach in computing the pK a and the free energies of formation of phosphate species in aqueous solution. Phys Chem Chem Phys 12:13804–13815. doi:10.1039/C0CP00175A
Tian L, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33:580–592
Xenides D, Randolf BR, Rode BM (2006) Hydrogen bonding in liquid water: an ab initio QM/MM MD simulation study. J Mol Liq 123:61–67. doi:10.1016/j.molliq.2005.06.002
Desiraju GR (2011) A bond by any other name. Angew Chem Int Ed 50:52–59. doi:10.1002/anie.201002960
Bader RFW (1990) Atoms in molecules—a quantum theory. Oxford University Press, Oxford
Ochterski JW (2000) Thermochemistry in Gaussian. http://www.gaussian.com/g_whitepap/thermo.htm, last accessed 15 Jan 2015
Paniagua C, Mota Valeri F (2010) Dipòsit Digital de la UB. U. de Barcelona, Barcelona. http://diposit.ub.edu/dspace/handle/2445/13382, last accessed 15 Jan 2015
Woon DE Jr, Dunning TH Jr (1995) The pronounced effect of microsolvation on diatomic alkali halides: ab initio modeling of MX(H2O) n (M = Li, Na; X = F, Cl; n = 1–3). J Am Chem Soc 117:1090–1097. doi:10.1021/ja00108a027
Lane JR, Contreras-García J, Piquemal J-P, Miller BJ, Kjaergaard HG (2013) Are bond critical points really critical for hydrogen bonding? J Chem Theory Comput 9:3263–3266. doi:10.1021/ct400420r
Elerman Y, Fuess H, Joswig W (1982) Hydrogen bonding in magnesium thiosulphate hexahydrate MgS2O3 ·6H2O. A neutron diffraction study. Acta Crystallogr B 38:1799–1801. doi:10.1107/S0567740882007225
Manojlović-Muir LA (1975) A neutron diffraction study of barium thiosulphate monohydrate, BaS2O3 ·H2O. Acta Crystallogr B 31:135–139. doi:10.1107/S0567740875002245
Llsensky GC, Levy HA (1978) Sodium thiosulfate pentahydrate: a redetermination by neutron diffraction. Acta Crystallogr B 34:1975–1977. doi:10.1107/S0567740878007116
Platts JA, Howard ST, Bracke BRF (1996) Directionality of hydrogen bonds to sulfur and oxygen. J Am Chem Soc 118:2726–2733. doi:10.1021/ja952871s
Dean JA (ed)(1985) Lange’s handbook of chemistry, 13th edn. McGraw-Hill, New York, pp 4–37, Table 4–4
Gimblett FGR, Monk CB (1955) Spectrophotometric studies of electrolytic dissociation. Part 1. Some thiosulphates in water. Trans Faraday Soc 51:793–802. doi:10.1039/TF9555100793
Acknowledgments
The authors wish to acknowledge funding from Universidad Autónoma de Nuevo León through the PAICyT program (grants #CN067-09 and #CA1731-07), and from Facultad de Ciencias Químicas.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 236 kb)
Rights and permissions
About this article

Cite this article
Rosas-García, V.M., Sáenz-Tavera, I. & Rojas-Unda, M. Microsolvation and hydration enthalpies of CaS2O3(H2O) n (n = 0–19) and S2O3 2−(H2O) n (n = 0–16): an ab initio study. J Mol Model 21, 98 (2015). https://doi.org/10.1007/s00894-015-2638-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-015-2638-9