
Abstract
Reaction mechanism for the benzaldehyde (ald) and 4-amine-4H-1,2,4-triazole (4at) has been investigated at the DFT (B3LYP)/6–31+G(d) computational level. Three transition states (TS) have been identified. The TS1 corresponds to hydrogen transfer from the NH2 group to the C = O bond and nucleophillic attack of the carbon atom from the aldehyde group on the nitrogen atom from the NH2 group in 4at. The result of this reaction is the hemiaminal molecule. The TS2 characterises an internal rearrangement of the benzene and triazole rings in the hemiaminal molecule. The TS3 leads to breaking of the O-H bond, the elimination reaction of the H2O molecule, and formation of the C=N bond. The final product of this reaction is a Schiff base. In order to determine the most favorable conditions for hemiaminal formation, the influence of electronic structure modification on the energetic properties during the reaction of benzaldehyde and 4-amine-4H-1,2,4-triazole has been studied. Thirteen substituents: NH2, OH, OCH3, CH3, F, I, Cl, Br, COH, COOH, CF3, CN, NO2, with different Hammett’s constant values (σ = −0.66–+0.78) have been considered. Finally, the reaction mechanism has been investigated in the presence of 1 to 5 water molecules.

The R substituent effect on the energetics of the reaction between (modified) benzaldehyde (R-ald) and 4-amine-4H-1,2,4-triazole
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
During the condensation reaction between primary amines and aromatic aldehydes, stable Schiff bases (imines) are formed through the interaction of functional groups and a water molecule elimination [1]. The reaction takes place in two stages. In the first one, an intermediate product, hemiaminal, is formed. It is characterized by the presence of a tetrahedral carbon atom, Ar-CH (OH) (NHR), and its general instability. During the second stage, a water molecule is eliminated, leading to a stable Schiff base, Ar-CH = NR, with the imine functional group. Schiff bases constititute a large group of stable and well known compounds. However, studies on hemiaminals, especially the open-chain ones, have been relatively rare due to their low stability. Hemiaminals have been found in solutions at low temperatures, [2, 3] or observed using sophisticated host-guest chemistry as trapped molecules in deep molecular cavities or in a porous coordination network [4–6]. From the NMR measurements, chemical half-lives of these compounds have been found to vary from 30 min to over 100 h at room temperature [7]. They have also been observed in solvent-free reactions as unstable compounds [8]. The first stable open-chain hemiaminal has been obtained in crystalline form from the reaction of di-2-pyridyl ketone with 4-cyclohexyl-3-thiosemicarbazide [9]. The compound was stabilized by the intramolecular hydrogen bonding.
Recently a new general method for an efficient preparation of stable open-chain hemiaminals has been proposed [10]. During synthesis, neutral solvent reaction conditions have been used and the reaction has been carried out using 4-amine-4H-1,2,4-triazole with benzaldehyde derivatives containing electron-withdrawing substituents (−NO2, −Cl), which decrease the electrophilicity of the carbonyl carbon atom. It has also been shown that using the same reactants in acidic conditions yields higher stability of a Schiff base.
The current paper focuses on the mechanism and energetics of the reaction between benzaldehyde (ald) and 4-amine-4H-1,2,4-triazole (4at) using ab initio calculations at the DFT (B3LYP) level. In order to find the best conditions improving the efficiency of the synthetic method and also to increase a range of compounds, the electronic structure of the reactants has been modified by varying the substituents and its effect on the reaction mechanism has been investigated. The substituents (R) with a wide range of the Hammett’s constants (σ) have been selected (−0.66–0.78). The following 13 R substituents have been chosen: NH2, OH, OCH3, CH3, F, I, Cl, Br, COH, COOH, CF3, CN, NO2. Initially, a monosubstituted derivative of benzaldehyde (R-ald) reacting with 4at has been studied. Then unsubstituted benzaldehyde (R=H, the reference molecule) and the 4at molecule reaction has been investigated with the 3,5 positions in the triazole ring subsequently occupied by a series of 13 R1 substituents (R1R1-4at): NH2, OH, OCH3, CH3, F, I, Cl, Br, COH, COOH, CF3, CN, NO2. Finally, the influence of varied number of water molecules (n = 1–5) on the reaction mechanism has been examined.
Computational details
The optimization of the geometrical structures and harmonic vibrational analysis has been performed using the GAUSSIAN 09 program [11]. The DFT (B3LYP) method [12–14] with the 6–31+G(d) basis set [15] as implemented in the Gaussian 09 program, has been used. The minima and transition states (TS) on the potential energy surface (PES) have been confirmed by the analysis of the harmonic vibrational frequencies. The reagents and products, connected by each TS, have been described using the IRC procedure [16, 17]. All calculations have been performed for the molecules in a singlet state.
Activation energies (Ea) have been calculated as a difference between the total energies (Etot) of the geometrical structures found on the IRC path toward the reagents (products) and subsequently optimized to local minimum of Etot and the total energy of TS. The vibrational zero-point energy difference (ΔZPVE) has been applied to all the Ea values, excluding one imaginary frequency for TS.
Interaction energies (Eint) for the pre-reactive (ald…4at) and post-reactive (Sch…H2O) complexes have been defined as a difference between Etot of the complex and Etot of the molecular fragments with geometrical parameters as in the respective optimized complex. The Eint values have been corrected (Eint CP) for the basis set superposition error (BSSE) using the counterpoise procedure (CP) [18]. Finally, ZPVE has been included in the values of Eint CP (Eint CP+ ΔZPVE).
Reaction energy (ΔEr) has been calculated as a sum of Etot of benzaldehyde and 4-amine-4H-1,2,4-triazole monomers and corrected for ΔZPVE.
All parameters, both geometrical and energetic, if not stated otherwise, have been calculated at the B3LYP/6–31+G(d) level of theory. Atom numbering in this paper corresponds to that adopted by Barys et al. in ref. [10].
Topological analysis of the electron density field, ρ(r) (AIM) has been carried out using AIMall program [19] at the DFT (B3LYP)/6–311++G(d,p) [20, 21] computational level. The following parameter for the bond critical points (BCP) are discussed: ρ(r) — the value of the electron density for BCP, Δρ(r) — the Laplacian value of the electron density for BCP, ε — “ellipticity” for BCP, H(r) — the total energy density for BCP.
Topological analysis of the electron localization function, η (r) (ELF) has been performed using TopMod package [22, 23] with a cubical grid with the step size of 0.08. The ELF domains have been visualized using the VMD program [24].
Benzaldehyde with 4-amine-4H-1,2,4-triazole reaction
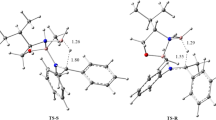
A search for transition states (TS), related to atomic rearrangement from benzaldehyde (ald) and 4-amine-4H-1,2,4-triazole (4at) to a Schiff base (Sch) and water (Scheme 1) yielded three TSs. The optimized geometrical structures of all stationary points are presented in Fig. 1. The TS1 describes the formation of the hemiaminal (t-hem1), TS2 describes internal reorientation of the triazole and benzene rings occuring in the equilibrium between two hemiaminal conformers (t-hem1 ↔ t-hem2) and the TS3 describes formation of a Schiff base and water. In the first reaction formation of hemiaminal starts with a pre-reactive complex from ald and 4at (ald…4at). In the second reaction, a post-reactive complex, formed between the Schiff base molecule and water (Sch…H2O) can be observed. The optimized geometrical structures of the ald…4at and Sch…H2O complexes are shown in Fig. 1.

The reaction between (modified) benzaldehyde and 4-amine-4H-1,2,4-triazole

The stationary points on PES and relative energies for the reaction of benzaldehyde (ald) with 4-amine-4H-1,2,4-triazole (4at) yielding the Schiff base (Sch) and water. The values of ΔE are calculated in realtion to the isolated ald and 4at molecules
Orientation of the C=O bond in relation to the NH2 group and the C-H bond from ald ‘pointing’ to the triazole ring nitrogen atom (see Fig. 1) suggests that the ald 4at complex is stabilized by intermolecular hydrogen bonds. Indeed, the topological analysis of the electron density, ρ(r) (AIM [25]) shows bond critical points (BCP) between the O1 atom from the C7 = O1 group and the H1 atom from the N4H1H2 group [ρ(r) = 0.019 e/bohr3, ρ(r) = 0.069 e/bohr5, ε= 0.040, H(r) = 0.002 Eh]. According to the Bader theory [26] those atoms are bonded and the respective stabilizing interaction can be associated with the N4-H1..O1 hydrogen bond. Similarly, the BCP [ρ(r) = 0.004 e/bohr3, ρ(r) = 0.014 e/bohr5, ε= 0.728, H(r) = 0.001 Eh] found between the C7-H3 bond from ald and the C2T atom of the triazole ring confirms the existence of the C7-H3…C2T interaction. Since the carbon atom plays a proton acceptor role here, this interaction is bound to be very week. All critical points for the ald…4at complex localized in the ρ(r) field are shown in Fig. 2a.

The critical points of index 0, 1, 2, and 3, localized for the electron density field in the intermolecular complexes formed between benzaldehyde and 4-amine-4H-1,2,4-triazole (ald…4at), and Schiff base and water (Sch…H2O)
The delocalization index, δ(A, B), defined as the integral of the exchange density over both atomic basins, A and B, is an important indicator of electron delocalization in the quantum theory of atoms in molecules. It is particularly important in bridging classical notions of bonding and quantum mechanics [27]. For the ald…4at complex, the value of δ (H3, C2T) index is about 0.01 and of δ(H1, O1) index is 0.06. This shows a very weak character of the H3…C2T interaction.
The interaction energy, Eint CP, for the ald…4at complex is −7.05 kcal mol−1 and the zero-point energy correction (ΔZPVE) changes its value to −5.42 kcal mol−1. The pre-reactive complex moves onto the TS1 after the supplied activation energy Ea I reaches the value of 45.49 kcal mol−1. The reaction is slightly endothermic and its product, hemiaminal (t-hem1) has energy 2.02 kcal mol−1 higher than the sum of the total energies (Etot) of ald and 4at. Calculated height of the activation barrier is large, thus the probability of spontaneous hemiaminal formation is very small. In experimental conditions other factors are bound to contribute to the decrease of the Ea I value. Solvation is one of the most possible ones, especially as water molecules are byproducts of the final reaction. The nucleophilic attack of the carbon atom from the C7 = O1 group occurs on the nitrogen, N4, from the NH2 group. This process is associated with proton transfer from the NH2 fragment to the O1 atom. There is no reason to expect that these two rearrangements occur at the same time on the reaction path.
Two mechanisms of this reaction can be considered and they are shown in Scheme 2. The former assumes proton transfer in the N4-H1…O1 bridge, followed by the C-N bond formation. The latter assumes the reverse. Since the formation of a covalent bond (C-N, O-H) requires transfer of the electron density from the carbon-oxygen bond into the interatomic region and the C7 = O1 bond has a polarized character with arbitrary partial charges, δ+ and 3δ−, on the C7 and O1 atoms, a simple analysis can be performed. If the proton transfer occurs first, the charge increase on the C7 atom (2δ+) facilitates the nucleophilic attack of N4 atom in the second step. If in contrast, the C-N bond formation occurs first, as in the second mechanism, the atomic charge decreases on the O1 atom (2δ−) and therefore the proton transfer is less favorable. This suggests that proton transfer in the N4-H1…O1 bridge facilitates the nucleophilic attack and should occur first.

Two mechanisms of electron density redistribution during the proton transfer and the C-N bond formation
In order to support this simple analysis, the electronic structure of the TS1 has been examined with the topological analysis of the ELF function [28–33]. Such bonding analysis is performed in a real space and the presence of particular valence bonding attractors indicates clearly the existence of covalent bonds [34, 35]. The ELF-localization domains for the TS1, related to topological basins of atomic cores, chemical bonds, and lone pairs are shown in Fig. 3. In the C7....N4 region, the valence bonding attractor, related to the covalent C7-N4 bond, has not been found. It is evident that the nucleofilic attack has not yet been finished and the new bond is not formed. In N4 vicinity, the ELF-domain corresponding to the lone pair is observed and its basin population is 1.73e. More advanced atomic rearrangement is observed within the valence region between the N4 and O1 atomic cores (N4....O1). The nitrogen-hydrogen (N4-H1) bond is already broken, confirmed by presence of the non-bonding domain on the N4 atom. The domain is related to the remains of the N4-H1 bond. Its basin population is 1.91e. The topology of η(r) function in the vicinity of detached H1 is described by the protonated asynaptic basin, V(H1). Such topological feature of the proton with the reminder electron density has been named “dressed proton” [36]. Its basin population is much larger than expected (0.51e). This result is similar to the one obtained for the proton transfer in malonaldehyde, 0.44e [36]. It is worth emphasizing that the O1-H1 bond in the TS1 is not formed at this stage. The detailed mechanism of the reaction between ald and 4at through the synthesis of hemiaminal and a Schiff base, has been studied lately using a combination of topological analysis of ELF and catastrophe theory [37]. The sequence of breaking and formation of chemical bonds obtained there, supports the results presented here. The first reactiom mechanism is as follows: the N-H bond in the 4at is broken, then the non-bonding basin V(O) on the O atom is created in ald, and finally the O-H bond is established. This atomic rearrangement is followed by nucleophillic attack of the carbon atom to nitrogen atom and a formation of the C-N bond.

The ELF-localization domains for TS1 and TS3
The product of the first reaction is the heminal molecule (t-hem1). Its optimized geometrical structure is presented in Fig. 1. The N3–N4–C7–C1 torsion angle, determining mutual orientation of the benzene and triazole rings is −74 [o]. The atomic chain is synclinal (sc) and both rings are approximately perpendicular. Barys et al. [10] described such conformation as twisted. In a stretched conformation, the chain is antiperiplanar (ap) and both rings are nearly parallel to each other. Among seven crystal structures, five hemiaminals with the stretched conformation and two with twisted conformation have been identified [10].
Formation of the Schiff base from hemiaminal requires elimination of a water molecule. This is only possible if the H2 atom and the lone pair on the O1 atom are in the positions supporting hydrogen transfer in the N4-H2…O1 bridge. Appropriate position of one of the O1 lone pairs in respect to the H2 atom is very important. Therefore, the molecule t-hem1 undergoes conformational change from a twisted conformation, where the N3–N4–C7–C1 torsional angle is −74 [o] (t-hem1), to a twisted conformation with the angle of 61 [o] (t-hem2). In t-hem1, the H2…O1 distance is 2.794 Å and is shortened to 2.559 Å in t-hem2. This process is associated with the transition state TS2 that describes internal equilibrium t-hem1 ↔ t-hem2. The N3–N4–C7–C1 torsion angle for the TS2 is −7 [o]. The activation energies, Ea III, Ea IV are smaller than 5 kcal mol−1. The reaction is slightly endothermic with the ΔEr of 3.80 kcal mol−1.
In the second reaction, a Schiff base is formed (TS3, see Fig. 1). The energetic barrier, Ea V, calculated from the direction of t-hem2 is 43.67 kcal mol−1. This result is 1.82 kcal mol−1 smaller than Ea I, thus the effectiveness of the Sch formation should be larger than that of hemiaminal. In respect to t-hem2, the reaction is slightly exothermic due to the value of Ea VI calculated in relation to the energy of the Sch…H2O complex, (44.70 kcal mol−1, Ea VI). Comparison with ald and 4at shows that the reaction occurs at an energy level which value is above the sum of their total energies.
In the reaction leading to the TS3, the C7-O1 bond is broken, the H2 atom is transferred to the O1 atom through the N4-H2…O1 bridge and the H2-O1 bond is created. Finally the water is separated from the Sch molecule. Furthermore, the single C7-N4 bond is transformed into the double C7 = N4 bond. Analysis of the bond lengths in the TS3 suggests that the N4-H2 bond is not broken at this stage. Furthermore, the presence of well isolated O1-H1 moiety implies a broken C7-O1 bond. In order to verify this statement, the topological analysis of η(r) function for TS3 has been carried out. The ELF-localization domains are presented in Fig. 3. The bonding attractor, V(N4, H2), can be found in the N4-H2 region. This confirms the presence of the N4-H2 bond. The C7-O1 bond is gone, which is confirmed by the absence of the V(C7, O1) attractor. The O1H1 fragment is described by the oxygen core basin, C(O1), the protonated H1-O1 bond basin V(H1, O1), and two non-bonding basins Vi=1,2(O1) related to two lone pairs in the Lewis formula. It is worth emphasizing that hydrogen transfer in the N4-H2…O1 bridge to one of the O1 lone electron pairs represented by Vi=1,2(O1) domains does not occur (see Fig. 4). Formation of the new H2-O1 bond requires a reorganization of the electron density in a valence shell of the O1 atom and as a result a new basin, V3(O1), is observed. In the reaction course that basin is joined to the V (H2) basin ‘creating’ the V(H2, O1) basin. Lone pairs, Vi=1,2(O1), observed in the geometrical structures of t-hem1, t-hem2 and TS3 are not directly involved in the formation process of the H2-O1 bond.

The ELF-localization domains for TS3. The lone pairs V1(O1) and V2(O1) are not involved in the proton transfer in the N4-H2…O1 bridge
The Sch and H2O molecules form a post-reactive complex, (Sch…H2O). The interaction energy Eint CP, ΔZPVE corrected, is −3.84 kcal mol−1. The optimized geometrical structure is shown in Fig. 1. Analysis of the interatomic distances suggests that the complex is stabilized by intermolecular hydrogen bonds. To verify this, the topological analysis of the ρ (r) field at the DFT (B3LYP)/6–311++G(d,p) level has been carried out. All localized CPs are shown in Fig. 2b. The BCPs have been found in the H2…N4 region (ρ(r) = 0.024 e/bohr3, Δρ(r) = 0.079 e/bohr5, ε= 0.074, H(r) = 0.002 Eh) and the H.N3 region (ρ(r) = 0.011 e/bohr3, Δρ(r) = 0.042 e/bohr5, ε= 0.158, H(r) = 0.002 Eh). The former can be associated with the O1-H2…N4 hydrogen bond formed between the water and Sch molecules. The latter describes the internal C2-H…N3 interaction, formed by the benzene ring hydrogen and the N3 lone pair. The value of delocalization index δ(H2, N4) is 0.072 and that of δ(H, N3) is 0.029, supporting much weaker character of the internal H…N3 interaction.
To sum up the conclusions in relation to the energetics: the two reactions and the internal equilibrium clearly proceed above the total energy level for both ald and 4at. The reaction energy, associated with the interaction of isolated water and Sch molecules, obtained in relation to ald and 4at, is 5.39 kcal mol−1. The whole process is therefore endoenergetic.
Reaction of R-monosubstituted benzaldehyde (R-ald) with 4-amine-4H-1,2,4-triazole
Effect of the benzaldehyde (R-ald) electronic structure modification on the reaction with 4-amine-4H-1,2,4-triazole energetics (Scheme 1) has been carried out for a series of 13 substituents (R = NH2, OH, OCH3, CH3, F, I, Cl, Br, COH, COOH, CF3, CN, NO2). The results have been compared with the results obtained for the reaction with the unsubstituted ald (R = H). Only para monosubstituted dervatives have been considered. It is widely recognized that the substituent electron-withdrawing and electron-donating properties are represented by the Hammett’s constant (σ) [38]. Table 1 shows clearly that the NO2 group, with the σp value of 0.78 has the maximal electron-withdrawing power. On the other hand, the NH2 group with σp of −0.66 has the maximal electron-donating power [38].
The mechanism of the reaction in question is similar to that of the reaction of ald with 4at (see Fig. 1). Three energy barriers are observed. Activation energies (Ea I, II, and Ea V, VI) are collected in Table 1. Furthermore, the interaction energies (Eint CP+ΔZPVE) for the pre-reactive complex (R-ald…4at) and the post-reactive complex (R-Sch…H2O) are shown. The final reaction energy, measured in relation to the isolated R-ald and 4at molecules, is represented by ΔEr.
In the discussion of the results we are going to concentrate on the activation energies only, Ea I and Ea V, associated with energy barriers in the first and second reaction. Activation energies are the most important parameters in the description of the hemiaminal and Schif base synthesis. The Ea I and Ea V, calculated for the unsubstituted ald and all 13 derivatives R-ald, are compared in Fig. 5. Firstly, Ea I and Ea V depend on the R substituent in different ways. In the case of Ea I the 0.93 kcal mol−1 range of calculated values is very small (see Fig. 5a). It is worth emphasizing that the barrier separating ald and 4at from t-hem1 does not change much with substitution. This is most probably a result of the complicated reaction mechanism (proton transfer and nucleophilic addition) combined with varied energy redistribution related to the change of substituent of different electron-withdrawing or donating properties. The largest value of Ea I has been obtained for ald modified with the CH3 group (45.60 kcal mol−1) and the smallest for NO2-ald (44.67 kcal mol−1). In the case of the barrier determining the formation of Sch, the 7.25 kcal mol−1 range of the Ea V values is much larger than those obtained for Ea I. The largest value (49.30 kcal mol−1) has been obtained for the substituent with the most electron-withdrawing group (NO2-ald), while the smallest (42.05 kcal mol−1) has been obtained for NH2-ald, where the substituent is known for its high electron-donating properties. For the barrier separating R-ald and 4at from hemiaminal, the modification of ald by ten R substituents (NO2, CN, COOH, COH, CF3, OCH3, Br, F, OH, I) results in the Ea I values smaller than that obtained for the unsubstituted ald. Thus any para-substitution of the ald molecule yields small but favorable effect in support of the synthesis of hemiaminal. The largest decrease of Ea I has been obtained for NO2, CN, COOH, COH, and CF3 groups. In the case of the Ea V barrier, the substitution yielded an increase in its value for all the substituents, except for the NH2 group yielding smaller value of the barrier in comparison to the barrier value obtained for the unsubstituted benzaldehyde (42.05 kcal mol−1 as opposed to 43.67 kcal mol−1). Thus, the effect is opposite and modification of ald does not facilitate the Schiff base formation. Only the NH2 substituent increases the efficiency of the Schiff base synthesis.

Comparison of the activation energy values, Ea I and Ea V illustrating the R substituent effect on the energetics of the reaction between (modified) benzaldehyde (R-ald) and 4-amine-4H-1,2,4-triazole. a) The values of Ea I and Ea V are arranged in ascending order, b) the relationship between the values of Ea I and Ea V and Hammett’s constant σp
Comparison of changes observed for the Ea I and Ea V barriers shows that modification of ald structure affects mainly the barrier determining formation of a Schiff base. The Ea I barrier characterizing the hemiaminal formation is relatively insensitive to the type of the R substituent. Thus, obtaining higher yield of the hemiaminal in its synthesis reaction through the modification of the ald electronic structure is presumably not caused by the height of Ea I decrease, but due to the increase of the Ea V barrier. From this point of view the NO2, CN, COOH, COH and CF3 substituents are the most desirable as they increase hemaminal formation efficiency through inhabiting the formation of a Sch (see Fig. 5a).
Figure 5b shows a relationship between Ea I and Ea V values, and the Hammett’s constant, σ. R substituents with increasing electron-withdrawing properties lead generally to a small decrease of the Ea I value. Analysis of the Ea V values shows an opposite trend and the R groups with an increasing σ value cause the barrier height increase. Correlation between Ea I, Ea V and σ analysed using linear regression is shown in Fig. 6. The determination coefficients (0.66, 0.89) indicate that the linear model works much better for the Ea V, represented by a wider range of the calculated values.

Analysis of the corelation between activation energy values, Ea I and Ea V, and the Hammett’s constant σp using linear regression
In order to ensure that our chosen level of calculations is correct, five R groups have been chosen, namely NH2, H, I, COH, and NO2 for the calculation with the 6-311++G(d,p) to see how higher level of calculations influences the activation energy values. The results of the Ea I, II and Ea V, VI are collected in Table 2. The basis set expansion leads to very small changes in the results. |ΔEa|ranges between 0.10 (R = NO2, Ea II) and 2.2 kcal mol−1 (R = COH, Ea VI). Essentially, the saturation of the basis set from 6-31+G(d) to 6-311++G(d,p) causes the maximum increase for the Ea VI value for the COH group, only by about 5 %. For the Ea I and Ea V values, studied in details here, the magnitude of change is smaller and ranges between 0.28 kcal mol−1 (R = COH, NO2) and 1.70 kcal mol−1 (R = I). We conclude therefore that the calculations performed at the B3LYP/6-31+G(d) level satisfactorlily describe energetic properties of the reaction between ald and 4at.
In the previous section a simple model for redistribution of the electron density (charge separation) during the reaction of ald and 4at confirmed by the ELF analysis has been proposed. That model predicted the increase of a positive charge on carbon as a consequence of the proton transfer. It is evident that such redistribution of the electron density must also be reflected by partial atomic charges obtained from a population analysis. In oder to verify this the approximation based on the atomic polar tensor (APT) proposed by Cioslowski et al. [39] have been used. Comparison of the APT charges calculated for the C7 and O1 atoms for ald and R-substituted aldehydes in the isolated molecules and the molecules reacting with 4at is shown in Fig. 7.

Comparison of the partial charges (in e) calculated for the C7 and O1 atoms in (modified) benzaldehyde (R-ald) for both optimized minimum energy structure (min, dotted line) and transition state (TS1, straight line)
For the C7 atom, in ald and R-ald molecules optimized to the energy minimum, almost all substituents increase its positive charge when compared to the unsubstituted ald (0.977e). The exceptions are the NO2 and CF3 groups that cause small decrease of the value of the APT charge (0.972e). The atomic charge values are within the range between 0.972e (NO2, CF3) and 1.107e (NH2). It is worth noting that the largest partial atomic charge for the C7 atom has been obtained for ald, modified with the NH2 group (1.106e) and the smallest one with CF3 and NO2 groups (0.972e). Similar analysis performed for the O1 atom shows that all R substituents with σ < 0 and the halogens (σ > 0) increase the value of the negative charge on O1 (−0.829e for NH2 - –0.747e for F) when compared to the unsubstituted ald (–0.742e). The substituents COH, COOH, CF3, CN and NO2 that lead to significantly smaller values of Ea I than those obtained for the unsubstituted ald (see Fig. 5) lead to the atomic charges smaller than those obtained for ald. It is worth noting that the largest atomic charge for the O1 atom has been calculated for ald modified with the NH2 group (−0.829e) and the smallest charge for the derivative with the NO2 group (−0.718e).
When the reacting system approaches TS, the atomic charges at the C7 and O1 atoms change their values (see Fig. 7). The C7 atom becomes more positively charged with the values varying from 1.319e for NH2 to 1.081e for NO2. Those extreme values correspond to the extreme values of the Hammett constant. However, the largest positive charge calculated for the C7 atom (NH2) does not correspond to the lowest value of the Ea I barrier. Similarly, the smallest positive charge calculated for the C7 atom (NO2) is found for the derivative with the smallest value of Ea I. In the case of the O1 atom almost all groups having σ > 0, with one exception of F atom, increase its negative charge in TS1 (−0.956e for I, –0.925e for H). The substituents having σ < 0 and F atom cause decrease of the negative charge.
It is interesting to examine the influence of the R group on the atomic charges for the H1 and N4 atoms in 4at. Those atoms are involved in interactions with the aldehyde group in the first reaction and the TS1. Respective values of the atomic charges, calculated with the APT method are shown in Fig. 8. For the optimized geometrical stuctures of the 11 derivatives (including 4at), very small range of values is observed. Such regularity is interrupted by exceptionally large values obtained for the COH and COOH groups (about 0.2e for H1 and −0.4e for N4). This can be caused by additional influence of internal hydrogen bonds formed between the N-H bonds from the NH2 group and the O atom from the COH and COOH groups. For the 11 derivatives mentioned the H1 atomic charge lies between 0.144e (CH3) and 0.183e (CF3), and for the N4 atom between −0.134e (OH) and −0.186e (I). After the reorganization for TS1 the values of atomic charge are much more perturbed than those observed for the minium structure (see Fig. 8). Nevertheless, for all the R1R1-4at derivatives the hydrogen atom H1 becomes more positively charged (0.329e for I - 0.480e for CH3) which can be caused by a electron density transfer in the O…H region. The charge on the N4 atom becomes less negative only for the OH and OCH3 groups but negative charge increase for other substituents can be found. It is worth noting that redistribution of the electron density, observed for the TS1 associated with hydrogen transfer increases both positive charge on the C7 atom and negative charge on the N4 atom (except for OH, OCH3) which facilitates nucelofillic addition. Unfortunately, the relationship between the substituent strength expressed by the Hammett constant and the TS1 activation energy cannot be easily obtained with the APT charges calculated for C7, O1 and H1, N4 atoms.

Comparison of the partial charge (in e) calculated for the H1 and N4 atoms in (modified) 4-amine-4H-1,2,4-triazole (R1R1-4at) for both optimized mínimum energy structure (min, dotted line) and transition state (TS1, straight line)
Reaction of benzaldehyde with R1R1-disubstituted 4-amine-4H-1,2,4-triazole (R1R1-4at)
The second way to influence barrier height in the formation reaction of the hemiaminal and Schiff base is to modify an electronic structure of 4-amine-4H-1,2,4-triazole. In order to perform this, 13 substituents (R1 = NH2, OH, OCH3, CH3, F, I, Cl, Br, COH, COOH, CF3, CN, NO2) in 3,5-positions have been selected (R1R1-4at). The Lewis formulas for the respective reactions are shown in Scheme 3. Similarly to the study on the modified benzaldehyde, only the first and second reaction have been selected due to their importance in hemiaminal (TS1) and Shiff base (TS3) formation.

The reaction between benzaldehyde and (modified) 4-amine-4H-1,2,4-triazole (R1R1-4at)
Activation energies for both reactions (Ea I, II, Ea V, VI) are collected in Table 3. For two substituents, NO2 and COH, we were unable to optimise the geometrical structures of the transition states. In the case of NO2, both TSs were difficult to localise. For the COH group only the TS3 could be found. Graphical comparison of the Ea I and Ea V values arranged in ascending order is shown in Fig. 9a.

Comparison the activation energy values, Ea I and Ea V illustrating the R1 substituent effect on the energetics of the reaction between benzaldehyde and (modified) 4-amine-4H-1,2,4-triazole (R1R1-4at). a) Ea I and Ea V values in ascending values, b) the relationship between the values of Ea I and Ea V and Hammett’s constant σp
In order to explain the reactivity trend for R1R1-4at, the influence of the R1 substituent on the electronic structure of 4at has been investigated. The calculations have been performed in real space using topological analysis of the ELF function. Similar investigations have been previously published for monosubstituted benzene derivatives [40], six-membered heterocycles [41], and monohalogen substituted phenols [42].
The electronic structure of unsubstituted 4at, represented by a set of the core and valence domains, corresponding to the ELF-basins (C(A) — core, V(A, B) — bonding and V (A) — non-bonding) is shown in Fig. 10. The basin populations \( \overline{N} \) for the C-N, C=N, N-N, N-H covalent bonds and formal lone pairs on the nitrogen atoms are presented in Table 4. The N-N and C=N covalent bonds are represented by single bonding basins with populations smaller than those expected on the basis of the Lewis formula. For example, formally double bonds, N2 = C2T and N1-C1T, are represented by the ELF-basins with the populations of 2.90 and 2.96e, respectively. The N-N bonds, formally single, are described by the basins with populations of 1.53e (N1-N2) and 1.37e (N3-N4). In contrast, the C-H, C-N and N-H bonds exhibit larger basin populations than the prediction for single bonds on the basis of Lewis formula. The ‘missing’ electron density is found mainly in the regions of the lone electron pairs. The lone pairs on the N1 and N2 atoms are described by the non-bonding localization basins with the populations of 3.04 and 3.02e, respectively. Similarly, the single non-bonding localization basin on the N4 atom exhibits a population of 2.18e. Due to the planarity of the triazole ring, the lone pair on the N3 atom is characterized by two ELF-basins, located below and above symmetry plane. The total population of two basins is 1.71e. It is worth noting that the lone pair on the N3 atoms has the smallest basin population among the lone pairs. This result can be explained by delocalization of its electron density in a five-membered ring.

The ELF-localization domains and the Lewis structure of 4-amine-4H-1,2,4-triazole
The topology of η(r) studied in the R1R1-4at derivatives is very similar to that in 4at except for the COH, COOH and NO2 derivatives. These groups have a strong delocalization ability within the triazole ring. As a result, its electronic structure is modified both in quantitative and qualitative aspects. Firstly, the local topology of η(r) in the valence space of the N3 atom is changed. The non-bonding electron density is characterized only by a single non-bonding basin, V(N3), with 0.70e (COH), 0.74e (COOH), and 0.81e (NO2), respectively. It is worth emphasizing that a basin population is greatly reduced in comparison to the formal 2e expected for a single lone pair. In comparison to the unsubstituted 4at, the electron density is shifted to the N3-C1T, N3-C2T, N1-N2, and N3-N4 bonds (see Table 3). For other derivatives, two non-bonding basins are found below and above the ring plane. Their total basin population is approximately two times larger than that of V(N3) and its value ranges from 1.68e (CN) to 2.10e (OCH3). It corresponds much better with a description of a single lone pair. Furthermore, such topology of ELF is similar to that observed for the unsubstituted 4at. Secondly, observed redistribution of the electron density causes the appareance of the new non-bonding basins V(C1T), V(C2T) in the valence space of the carbon atoms C2T. Their basin populations are relatively small: 0.21 and 0.09e for the COH derivative and 0.33, 0.03e for the COOH derivative. Interestingly, no such basins are observed for the NO2 derivative. Although both V(C1T) and V(C2T) basins are unexpected topological features, they emphasize redistribution of the electron density to the triazole ring.
Correlation between the difference of the Hammett’s constants for meta and para positions (σp-σm) and the population of the bonds and lone pairs in 4at and R1R1-4at derivatives are presented in Fig. 11. The linear regression with the correlation coefficient larger than 0.5 has been found only for the N1-N2 and N3-N4 bonds and the lone pairs on the N1 and N2 atoms. In the case of the N1-N2 bond of the triazole ring, the R1 groups with increasing electron-withdrawing properties cause the depopulation of the bond. Similar effect is observed for the N3-N4 bond, although the correlation is much smaller. The depopulation of the N1-N2 bond leads to saturation of the non-bonding basins corresponding to the lone pairs on the N1 and N2 atoms. For other bonds and lone pairs no essential linear correlation between the differences σp-σm and the basin populations is observed.

Relationship between the basin population, \( \overline{N} \) [e] of the chemical bonds and lone pairs in (modified) 4-amine-4H-1,2,4-triazole (R1R1-4at), localized using topological analysis of electron localization function (ELF) and the difference of the Hammett’s constants for the meta and para positions (σp-σm)
Various substituents in 4at influence the energetic profile of the reaction between ald and R1R1-4at. Comparison of the Ea I and Ea V values (see Figs. 9a and b) shows that for the OH, OCH3, F, I, Br, Cl, COOH, CN, and CF3, groups, the Ea V barrier, determining formation the Schiff base (TS3) is larger than the Ea I. This is a favorable situation for the kinetic stabilization of hemiaminal. Only the Br and F substituents yielded slightly higher Ea I values (0.36 and 0.89 kcal mol−1) than calculated for unsubstituted 4at. Thus, other substituents (CH3, I, Cl, CF3, OH, OCH3, NH2, CN, COOH) can be used in order to increase the hem formation probability. The largest decrease in the height of Ea I was obtained for the COOH, CN, NH2, OCH3, and OH groups: 5.81, 4.65, 4.04, 2.83, and 2.12 kcal mol−1, respectively. In the case of the Ea V barrier determining the Schiff base formation, all substituents except the NH2 group increase the barrier height. Thus all these substituents are favorable for the hem stabilization as they decrease probability of a Schiff base formation. Comparison of the results obtained for both TSs, shows that the OCH3 and OH groups seem to be the most effective. The respective energy barriers are relatively high for Ea V (48.11, 47.53 kcal mol−1) and relatively small for Ea I (42.66, 43.37 kcal mol−1) compared to the reaction with the usubstitued 4at (45.49, 43.67 kcal mol−1).
Figure 9b presents Ea I and Ea V plotted against the increasing difference of the Hammett’s constants for the meta and para positions (σp-σm). A difference of the σ values has been used to establish a positional hierarchy of the substituents. It is evident that modification of 4at results in the activation energy changes for a whole range of the σ values, but lack the regularity observed for the reaction with the unsubstituted 4at (see Fig. 5). Therefore the application of the σ parameter for molecules with similar structure to 4-amine-4H-1,2,4-triazole seems to be questionable.
The influence of water solvation on the reaction of benzaldehyde with 4-amine-4H-1,2,4-triazole
Experimental conditions, under which the hemiaminal synthesis is usually conducted and the fact that the Schiff base formation is associated with the water molecule release, suggest solvent effect importance on the energy properties related to the reaction between benzaldehyde and 4-amine-4H-1,2,4-triazole. The microsolvation process has been studied in the gas phase. Water molecules (1–5) have been placed around the reagents involved (ald, 4at, hem, Sch) and the obtained systems underwent geometry optimizations. Optimized geometrical structures of the substrates, TS1, and the products associated with the hemiaminal formation in the presence of up to five water molecules are shown in Fig. 12. Similar research has been published by Williams [43] for the addition reaction (ammonia with formaldehyde) and Hall and Smith [44] for the reaction between methylamine and formaldehyde.

Optimized geometrical structures for the TS1 in the reaction of benzaldehyde with 4-amine-4H-1,2,4-triazole in the presence of one and five H2O molecules
Analysis of the geometrical structures at the TS1 shows that the presence of one water molecule, results in the formation of two intermolecular hydrogen bonds, O-H…O and N-H…O. In the molecular clusters constructed this way, double proton transfer is observed. The hydrogen atom from the NH2 group is transferred to the water molecule while the oxygen and the hydrogen atom from the H2O molecule is shifted to the C=O bond. Finally, the O-H bond is formed and the water molecule is released. Similar mechanisms are observed for two, three, four, and five water molecules. In the case of TS3, one H2O molecule participates in the intermolecular hydrogen bonds, O-H…O and N-H…O, for the systems up to two water molecules. When the number of water molecules increases to three, four or five, intermolecular hydrogen bonds are formed by two H2O molecules (see Fig. 13).

Optimized geometrical structure for the TS3 with three molecules of water
Comparison of the barrier heights, Ea I and Ea V, calculated for an increasing number of water molecules (see Fig. 14) shows a dramatic decrease of the activation energy. The largest effect is found for the barrier separating the hem from ald and 4at with three H2O molecules surrounding the reacting molecules, because the Ea I is reduced by approximately 57 % (25.79 kcal mol−1). For the barrier controlling the formation of the Schiff base, Ea V, the largest effect is found when five water molecules are involved. The Ea V is reduced by approximately 38 % (16.72 kcal mol−1). It is worth emphasizing that solvent effect is much more profound during formation of hemiaminal than during formation of Sch. These results confirm that solvent effects largely influence the conditions under which the hemiaminal is formed.

Change in the activation energies, Ea I and Ea V determining the energetics of the reaction between benzaldehyde and 4-amine-4H-1,2,4-triazole in the presence of 1 to 5 water molecules
Conclusions
Quantum-chemical calculations using DFT (B3LYP) method have been performed to determine important factors in the process of hemiaminal synthesis and stabilization. The reaction mechanism of benzaldehyde and 4-amine-4H-1,2,4-triazole synthesis to form hemiaminal and subsequently leading to a Schiff base has been proposed and studied. A set of 13 substituents of various electron-withdrawing and electron-donating properties has been examined, with main focus on the substitution effect on the electronic structure and energetic properties of the substituents, namely benzaldehyde and 4-amine-4H-1,2,4-triazole and the whole reaction process. The results aid devising the reaction mechanism of the hemiaminal and Schiff base formation and specifying the nature of favorable substituents in hemiaminal synthesis. The main findings are presented below:
-
1.
The mechanism of reaction between the benzaldehyde and 4-amine-4H-1,2,4-triazole consists of two steps.
-
2.
First, the proton transfer in the N-H…O bridge occurs. The proton (dressed proton) is transferred from 4-amine-4H-1,2,4-triazole to benzaldehyde and this process is followed by the nucleophilic attack of the carbon atom from the COH group on the nitrogen atom in the 4-amine-4H-1,2,4-triazole. The product of this reaction is a twisted conformation of the hemiaminal molecule. The reaction is preceded by an intermolecular complex formation between the ald and the 4at. Similar mechanism was described by Feldman et al. [45] for the reaction of ammonia with formaldehyde and Erdtman et al. [46] for a range of systems (acetone, aminoacetone plus methylamine) that modeled the active site of porphobilinogen synthase.
-
3.
Next, the internal equilibrium occurs between two conformers of the hemiaminal molecule. The distance between the H atom of the N-H bond and the O atom of the O-H group decreases from 2.794 to 2.559 Å. It is worth noting that a similar step (TS2) was also observed by Erdtman et al. [42] and described as “low barrier carbinolamine rearangement”.
-
4.
The second step is the C-O bond breaking, followed by internal proton (dressed proton) transfer in the N-H…O bridge. The product of the reaction is an intermolecular complex, formed between Schiff base molecule and water.
-
5.
A simple model of atomic rearrangements, associated with the TS1 based on the charge separation in the C=O bond and electron density redistribution during the formation of new bonds, suggests that the proton transfer facilitating nucleophilic attack of the C atom on the N atom takes priority. Topological analysis of the ELF function, carried out for the TS1, supports this model, showing a broken N-H bond in the absence of the C-N bond. This has also been confirmed before by detailed topological analysis of ELF combined with the catastrophy theory [37].
-
6.
Modification of the electronic structure of benzaldehyde using a variety of R substituents with different electron-withdrawing and electron-donating properties has a very small influence on the height of the energetical barrier Ea I separating the ald and 4at from hemiaminal. The influence of the substitution is much more profound on the barrier, Ea V, related to the Schiff base formation. Stabilization of the hemiaminal, the main product of the reaction, occurs mainly as a result of the Ea V barrier height increase.
-
7.
Modification of the benzaldehyde structure using the NO2, CN, CF3, COH and COOH, substituents gives the most significant effects manifested in the Ea I barrier height decrease with a simultaneous increase the Ea V barrier height.
-
8.
Modification of 4-amine-4H-1,2,4-triazole with almost all R1 substituents results in a favorable stabilization of hemiaminal, decreasing the height of Ea I and increasing the height of Ea V. The only exceptions are the Br and F substituents as they increase the height of Ea I slightly when compared to the usubstituted 4at. On the other hand, the NH2 group reduces the height of the Ea V barrier.
-
9.
Solvent effect drastically reduces the barrier height for the hemiaminal formation. When three water molecules are considered, Ea I is decreased by about 57 %. For the Schiff base formation, the effect is smaller. When five water molecules are involved, Ea V is decreased by about 38 %.
References
Smith JMB, March J (2007) March’s advanced organic chemistry. Wiley Interscience, Wiley, 6th edn, pp 1281–1282
Forlani L, Marianucci E, Todesco PE (1984) J Chem Res 1984:126
Chudek J (1985) A. Foster, R. Young, D. 13C nuclear magnetic resonance studies of the products of reaction of acetaldehyde and of simple ketones in liquid ammonia, in hydrazine hydrate, and in some substituted hydrazine solutions. J Chem SocPerkin Trans 2:1285–1289
Iwasawa T, Hooley RJ, Rebek J Jr (2007) Stabilization of labile carbonyl addition intermediates by a synthetic receptor. Science 317:493–496
Hooley RJ, Iwasawa T, Rebek J Jr (2007) Detection of reactive tetrahedral intermediates in a deep cavitand with an introverted functionality. J Am Chem Soc 129:15330–15339
Kawamichi T, Haneda T, Kawano M, Fujita M (2009) X-ray observation of a transient hemiaminal trapped in a porous network. Nature 461:633–635
Forlani L, Marianucci E, Todesco PE (1984) H-1 nuclear magnetic-resonance evidence for tetrahedral intermediates in the reactions between aromatic carbonyl groups and aliphatic-amines. J Chem Res-S 4:126–127
Dolotko O, Wiench JW, Dennis K, Pecharsky W, Balema VK (2010) Mechanically induced reactions in organic solids: liquid eutectics or solid-state processes? New J Chem 34:25–28
Suni V, Kurup MRP, Nethaji M (2005) Unusual isolation of a hemiaminal product from 4-cyclohexyl-3-thiosemicarbazide and di-2-pyridyl ketone: structural and spectral investigations. J Mol Struct 749:177–182
Barys M, Ciunik Z, Drabent K, Kwiecien A (2010) Stable hemiaminals containing a triazole ring. New J Chem 34:2605–2611
Gaussian 09, Revision D.01, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA et al. (2009) Gaussian Inc, Wallingford, CT
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 38:3098–3100
Becke AD (1993) Density‐functional thermochemistry. III The role of exact exchange. J Chem Phys 98:5648–5652
Lee CT, Yang WT, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Frisch MJ, Pople JA, Binkley JS (1984) Self‐consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J Chem Phys 80:3265–3269
Fukui K (1970) Formulation of the reaction coordinate. J Phys Chem 74:4161–4163
Fukui K (1981) The path of chemical reactions - the IRC approach. Acc Chem Res 14:363–368
Boys SF, Bernardi F (1970) The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys 19:553–566
Todd A, Keith TK (2013) AIMAll (Version 12.11.09). Gristmill Software, Overland Park, KS (aim.tkgristmill.com)
Krishnan R, Binkley JS, Seeger R, Pople JA (1980) Self‐consistent molecular orbital methods. XX A basis set for correlated wave functions. J Chem Phys 72:650–654
Clark T, Chandrasekhar J (1983) Schleyer, P.v.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li–F. J Comp Chem 4:294–301
Noury S, Krokidis X, Fuster F, Silvi B (1997) TopMod, Paris
Noury S, Krokidis X, Fuster F, Silvi B (1999) Computational tools for the electron localization function topological analysis. Comput Chem 23:597–604
Humphrey W, Dalke A, Schulten K (1996) VMD: Visual molecular dynamics. J Mol Graphics 14:33–38
Bader R (1994) Atoms in molecules: a quantum theory. Oxford University Press, USA
Bader R (1991) A quantum theory of molecular structure and its applications. Chem Rev 91:893–928
Fradera X, Austen MA, Bader R (1999) The Lewis model and beyond. J Phys Chem A 103:304–314
Becke AD, Edgecombe KE (1990) A simple measure of electron localization in atomic and molecular systems. J Chem Phys 92:5397–5403
Silvi B, Savin A (1994) Classification of chemical bonds based on topological analysis of electron localization functions. Nature 371:683–686
Savin A, Silvi B, Colonna F (1996) Topological analysis of the electron localization function applied to delocalized bonds. Can J Chem 74:1088–1096
Silvi B (2002) The synaptic order: a key concept to understand multicenter bonding. J Mol Struct 614:3–10
Savin A (2005) The electron localization function (ELF) and its relatives: interpretations and difficulties. J Mol Struct (THEOCHEM) 727:127–131
Savin A (2005) On the significance of ELF basins. J Chem Sci 117:473–475
Chevreau H, Fuster F, Silvi B (2001) La liaison chimique: mythe ou réalité. Les méthodes topologiques de description de la liaison. L’Actual Chim 240:15–22
Silvi B, Fourre I, Alikhani ME (2005) The topological analysis of the electron localization function. A Key for a position space representation of chemical bonds. Monatsh Chem 136:855–879
Krokidis X, Goncalves V, Savin A, Silvi B (1998) How malonaldehyde bonds change during proton transfer. J Phys Chem A 102:5065–5073
Berski S, Ciunik LZ The mechanism of the formation of the hemiaminal and Schiff base from the benzaldehyde and triazole studied by means of the topological analysis of electron localization function and catastrophe theory. Mol. Phys. DOI: 10.1080/00268976.2014.974702
Hansh C, Leo A, Taft RW (1991) A survey of Hammett substituent constants and resonance and field parameters. Chem Rev 9l:165–195
Cioslowski J (1989) A new population analysis based on atomic polar tensors. J Am Chem Soc 111:8333–8336
Fuster F, Sevin A, Silvi B (2000) Topological analysis of the electron localization function (ELF) applied to the electrophilic aromatic substitution. J Phys Chem A 104:852–858
Fuster F, Sevin A, Silvi B (2000) Determination of substitutional sites in heterocycles from the topological analysis of the electron localization function (ELF). J Comp Chem 21:509–514
Silvi B, Kryachko ES, Tishchenko O, Fuster FT, Nguyen M (2002) Key properties of monohalogen substituted phenols: interpretation in terms of the electron localization function. Mol Phys 100:1659–1675
Williams IH (1987) Theoretical modelling of specific solvation effects upon carbonyl addition. J Am Chem Soc 109:6299–6307
Hall NE, Smith BJ (1998) High-level ab initio molecular orbital calculations of imine formation. J Phys Chem A 102:4930–4938
Feldmann MT, Widicus SL, Blake GA, Kent DR IV, Goddard WA III (2005) Aminomethanol water elimination: theoretical examination. J Chem Phys 123:034304–1
Erdtman E, Bushnell EAC, Gauld JW, Eriksson LA (2011) Computational studies on Schiff-base formation: implications for the catalytic mechanism of porphobilinogen synthase. Comput Theor Chem 963:479–489
Acknowledgments
The authors are grateful to the Wroclaw Centre for Networking and Supercomputing for generous allocation of computer time.
The research was supported by Wroclaw Research Center EIT+ under the project “Biotechnologies and advanced medical technologies — BioMed” (POIG 01.01.02-02-003/08-00) financed from the European Regional Development Fund (Operational Program Innovative Economy, 1.1.2).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Berski, S., Gordon, A.J. & Ciunik, L.Z. The DFT study on the reaction between benzaldehyde and 4-amine-4H-1,2,4-triazole and their derivatives as a source of stable hemiaminals and schiff bases. Effect of substitution and solvation on the reaction mechanism. J Mol Model 21, 57 (2015). https://doi.org/10.1007/s00894-015-2606-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-015-2606-4