
Abstract
Human dopamine D4 receptor is a GPCR target in the treatment of neurological and psychiatric conditions such as schizophrenia and Parkinson’s disease. The X-ray structure of this receptor has not been resolved so far. Therefore, a proper 3D structure of D4 could provide a good tool in order to design novel ligands against this target. In this study, homology modeling studies were performed to obtain a reasonable structure of the receptor using known templates. The obtained model was subjected to molecular dynamic simulation within a DPPC membrane system. Some structural features of the receptor such as a conserved disulfide bridge and ionic lock were considered in the modeling experiments. The resulted trajectories of simulation were clustered based on the root mean square deviation of the backbone. Some known ligands and decoys were accordingly docked into the representative frames of each cluster. The best final model was finally selected based on its ability to discriminate between active ligands and inactive decoys (ROC = 0.839). The presented model of human D4 receptor could be a promising starting point in future studies of drug design for the described target.

Superposition of human D4 model with the crystal structure of D3 at TM regions
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
G protein coupled receptors (GPCRs), as a large family of transmembrane proteins, have key functions in receiving extracellular signals and physiological function of the body [1]. Therefore, they are important targets in a wide spectrum of diseases [2]. In terms of therapeutic relevance, approximately 500 members of this family have been identified, although only a few of them were structurally characterized [3].
Dopamine receptors, as one group of the GPCRs family, are best known for their regulating roles in essential functions including cognitive processes and emotional status [4]. Adverse activation of dopaminergic pathways takes part in neuropathological disorders such as schizophrenia, Parkinson’s disease, bipolar disorder, attention deficit hyperactivity disorder (ADHD), Huntington’s disease, and Tourette’s syndrome [5]. Among dopamine receptors, D4 is mostly expressed in GABAergic neurons of the cerebral cortex, hippocampus, and substantia nigra [6]. Therefore, D4 receptor plays an important role in cognitive tasks and memory performance and is one of the major goals in treating schizophrenia [7]. Some efforts were based on synthesis of selective ligands against D4 receptor. However, most of these compounds indicated weak affinities toward D4 receptor [8, 9]. ML398 has been presented as a selective D4 antagonist in recent studies [10]. Moreover, some selective ligands exhibited remarkable binding affinity to 5HT1AR ,5HT2AR, and 5HT2BR [11, 12]. A SAR study of selective D4 ligands reported an important arene-cation interaction between the ligands arene moiety and the residue Arg-186. This interaction was found to be critical in designing potent D4 ligands [13]. Also a ligand-based QSAR study for the antagonists of human D4 was reported in the literature [14]. According to these efforts, it is of great importance to get knowledge about D4R three dimensional (3D) structure. Obtaining 3D structure of transmembrane proteins is a difficult task as they are embedded in the lipid membrane and extracting adequate amount for X-ray crystallography is hard to achieve [15]. In addition, lipid membrane can affect protein conformation so that obtaining a true conformation by empirical method is so challenging [16]. Despite their importance, transmembrane protein structures constitute only 1 % of the Protein Data Bank [17]. Recently, 3D structures of some GPCRs, such as human dopamine D3 receptor (D3R), have been reported by experimental methods [18]. Since dopamine receptors are to some extent similar with regard to their sequences, human D3R structure can be utilized as the main template for homology modeling of other dopamine receptors such as D4R.
More knowledge about the structural features of human D4R could lead to progress in the area of designing potent and selective compounds against this target. A reasonable approach to reach a 3D conformation of the receptor for further binding mode analysis is molecular modeling methods [19]. Homology modeling is a technique able to identify and refine models of the receptor. It can also provide valuable information for further structure based virtual screening studies [20, 21].
In this study, a predictive 3D structure of D4R was proposed by means of homology modeling technique. MD studies on the whole system (protein in DPPC membrane) were also carried out to equilibrate and optimize the protein in physiological conditions [22, 23]. Subsequently, the resulted frames were evaluated for their ability to distinguish between experimentally known ligands and decoys of the receptor using docking approach. The frame with a higher predictive ability was finally proposed as a model for the natural conformation of the receptor at the binding site. Results of this context could possibly lead to structure-based drug design and targeting of this receptor in future studies [24].
Materials and methods
Homology modeling studies
The primary sequence of human dopamine receptor type 4(DRD4, access number P21917) was retrieved from UniProt database at www.expasy.org [25]. The FASTA sequence was subjected to Basic Local Alignment Search Toolbox (BLAST) using BLOSUM62 scoring matrix at NCBI database [26]. Recursive runs of BLAST were repeated for transmembrane regions of the receptor to find more related templates for these regions. Finally, four templates [PDB code: 3pbl (humandopamine D3 receptor in complex with Eticlopride), 3UON(human M2 muscarinic acetylcholine receptor bound to an antagonist), 4GRV(the neurotensin receptor NTS1 in complex with neurotensin), and 4IB4 (the chimeric protein of 5-HT2B-BRIL in complex with ergotamine)] were obtained based on their similarity toward D4 receptor sequence. Subsequently, a multiple alignment file has been generated between the sequence of the receptor and the templates using ClustalX2 software [27]. Some modifications were done in the alignment file by manual insertion and removing of the gap penalties.
For prediction of transmembrane helices, different methods including HMMTOP [28], TMHMM [29], DAS [30], SOSUI [31], TMpred [32], PolyPhobius [33], predict protein [34], APSSP [35], ExPASY [25], and TOPCONS [36] were employed. The final alignment file was used for 3D generation of D4 receptor using modeler 9v12 software [37]. About 100 different structures of the receptor were modeled with regards to energy refinement as implemented in modeller9v12 [38]. An in-house application (Modelface) was used, as interface to Modeller software, for generation and running of the python scripts needed for the experiments. A conservative disulfide bridge was introduced in all models between the residues 108 (extra cellular loop 2) and 185 (extra cellular loop 3). Ranking of the resulted models was done based on DOPE score values generated by modeler software. Afterward, Bioinf, Tasser, and Falc servers were utilized to refine the models and enhance the accuracy of low conserved loop regions [39–41]. The model with the highest DOPE score value was submitted to Procheck server for further verification [42].
MD simulation
MD simulation study was performed on the final model obtained from homology modeling studies. The protein PDB structure was embedded in a DPPC membrane at appropriate orientation and location of TM regions. Before embedding the PDB structure, all coordinates were translated to proper locations based on the data of TM regions. Different algorithms have been presented to equilibrate a receptor inside lipid membrane. A very useful and convenient method with less consumption of time during equilibrium is called InflatGRO [43]. Therefore, to equilibrate the resulted system of lipid-protein, InflateGRO perl script was used [43]. At first, lipids and protein were placed on an expanded grid and then shrinking process was done consecutively to reach the favorable density area per lipid for DPPC membranes (62.9–64 Å2) [44]. GridMAT-MD_v2.0 perl script was used to calculate area per lipid of the resulted system [45]. The system was then solvated with spc216 water model and a concentration of 0.15 M NaCl was inserted to simulate the physiological conditions. Energy minimization followed by two short runs of MD (1 ns) was done on the whole system to stabilize it for main run of simulation. Steepest descent minimization integrator with maximum 50,000 cycles and maximum 1000.0 kJ (mol nm)−1 force were used during minimization. NVT and NPT ensembles with restrains on protein backbone were used for two runs of MD. The main run of MD was subsequently carried out and the system and trajectories were subjected to further studies. Leap-frog integrator with LINCS constraint algorithm and particle mesh Ewald coulomb type were set for MD. The more accurate Nose-Hoover thermostat together with Parrinello-Rahman pressure coupling method was used during the main run of MD. Energy and RMSD plot calculations and visualizations of the structures were done using Gromacs and VMD softwares, respectively [46]. All MD experiments were done using GROMOS force field implemented in Gromacs 4.5.3.[47].
Docking studies
Q-site finder was used in order to find the potential binding sites of the receptor [48]. Subsequently, different frames of the receptor were extracted after clustering MD trajectories using principal component analysis. For this purpose, a vector of backbone variation for all residues within a frame was generated by means of VMD software. The resulted matrix for all frames was subjected to singular value decomposition (SVD) algorithm as implemented in MATLAB software (Mathworks Inc). The clusters were obtained based on the plot of the first two PCs with maximum eigenvalues. Two frames inside each cluster were selected for further docking studies. In docking simulation study, 121 small molecule structures with experimental biological activities were obtained from EMBL database as SMILES [49]. Based on their IC50 values, 31 active structures and 90 inactive compounds were considered as ligands and decoys, respectively.
3D generation and energy minimization of the structures were accomplished using Openbabel 2.3.2 [50]. Docking simulations was performed using Autodock4.2 and grid box was centered on Cα atom of Asp115 with 60×60×60 dimensions. Parameters of genetic algorithm were set to ten runs, 2,500,000 energy evaluations and 150 population size.
The docking scores of ligands and decoys were analyzed by receiver operating characteristic (ROC) curves [51]. In addition, all visual inspection of protein ligand complexes was done using VMD software [46].
Results and discussion
To achieve an optimized 3D structure for D4 receptor, homology modeling was used in order to predict the 3D structure of receptor using known similar proteins. The templates used in modeling of the receptor are listed in Table 1. 3PBL (D3) was used as the main template due to its higher identity toward human dopamine D4 receptor (31.54 %). Helices orientations and diversity in loop portion are the main structural difference between D2-like receptors. Length of the sequence in human D4 (467) differs from that of D3 (400). It is also worth noting that 3PBL, 3UON, and 4GRV are nearly 50 % identical to each other in terms of their primary sequence. As described earlier, the homologue templates were found by means of basic local alignment search tool (BLAST) at NCBI and the threading based method as implemented in Tasser server. As displayed in Fig. 1, the sequences were aligned in such a way to cover the maximum area of the sequence for dopamine receptor. The alignment procedure was the key stone in homology modeling of the receptor. Since there was not any favored template covering the first extracellular loop (residue 1–37), Falc server was used to model and refine this part of the protein. Using refinement step led to dramatic enhancement of the final model. Different techniques could be applied for quality validation of the models. In this study, the primary ranking of the generated models was based on DOPE score according to the energy of the final 3D structures. Moreover, the final model needed to be subjected for further validations [42]. Ramachandran plot, as the output of PROCHECK software, assessed the final quality of protein by indicating residues located in disallowed regions and percentage of those in appropriate zone [42]. As displayed in Fig. 2, there are few residues located in disallowed regions and more than 96 % of residues are found in favored or allowed regions of the plot. It is therefore clear that the resulted model meets the required quality criteria for further studies.

The final alignment used in homology modeling step

Ramachandran plot of human dopamine D4 structure
Natural environment of body and cell membrane would cause conformational change in target protein. One useful method for transformational prediction, which makes a 3D model adapt with its physiological environment, is molecular dynamic simulation. TM regions of the receptor must be embedded in lipid bilayer at proper orientation before running simulation studies of a membrane protein. For this purpose, different methods described in the methodology section were aimed to predict transmembrane (TM) regions of the receptor. As displayed in Table 2, all methods converged reasonably in predicting TM residues of the receptor.
GPCRs and other membrane bound proteins are inserted into phospholipid membranes following a very specific geometry and according to their particular chemistry called “amphipathic character” (polar heads of phospholipids are in contact with water and hydrophobic aminoacids of membrane bound proteins in close contact with the hydrophobic tail of phospholipids). For this purpose the receptor was translated with care at correct orientation inside the membrane. As depicted in Fig. 3, the obtained geometry was in accord with the data of Table 2. The system was solvated in such a way not to permit penetration of water molecules inside hydrophobic TM regions of the receptor. To evaluate the conformational stability of the predicted protein during MD simulation, root mean square deviation (RMSD) plot was calculated on the protein backbone. As shown in Fig. 4, the simulation was going on 50 ns from the starting point. The energy plot revealed a stable status for the resulted system during simulation. In this study, heat map analysis of Cα RMSD was used for showing conformational changes within the system during simulation. The results of heatmap for all trajectories are displayed in Fig. 5. The most conformational fluctuations occurred in residues 1–32 and 240–350 which are located in the first extracellular and third cytoplasmic domains, respectively. In residues 1–32, the N-terminal of the protein is unrestrained so its conformation was prone to variations. In case of cytoplasmic domain (ICL3, residues 240–350), its nature as a long sequence, led to extra flexibility for this region and more conformational fluctuations were therefore anticipated.

The correct orientation of the receptor within lipid bilayer based on the data displayed in Table 2

RMSD of Cα during 50 ns MD simulation (left), Energy plot of MD simulation (right)

Heatmap analysis of trajectories during MD simulation
The conserved disulfide bridge between the two residues 108 in EL2 and 185 in EL3 was also deterministic in molecular dynamic simulation (Fig. 6). A similar disulfide bridge was previously reported in the models of human dopamine D2 receptor as well as in the crystal structure of D3 [18, 52]. However, in D4, disulfide bridge is located between ECL2 (108) and ECL3 (185) but in case of D3 receptor the location is at ECL2 (Cys 181) and helix 3 (Cys103) [18].

Presence of ionic lock and disulfide bridge in D4 model as two structural features of the receptor
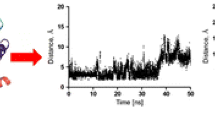
A common feature of most class A GPCRs is the presence of ionic lock between “D[E]RY” motif and a negatively charged residue at the front cytoplasmic domain [18, 53]. Since our model was to represent the receptor at antagonist state, it was of great importance to monitor the formation of the aforementioned salt bridge between cytoplasmic region of helices III (Arg133) and IV (Glu389) (Fig. 6). The nature of ionic lock across NH chain of Arg133 and OE of Glu389 was investigated using the distance calculations for these two residues. As seen in Fig. 7, the ionic lock was present at steady state of the simulation.

Distance calculations between Arg133N and Glu389O. Ionic lock could be observed at steady states of the simulation
Docking studies were used as a tool for further validation of the experiments and to find a useful model for future studies of drug design. To avoid possible errors caused by random selection of the representative frames, we used an unsupervised classification approach. Two representative frames were thereafter selected within each cluster of PCA for docking purpose (Fig. 8).

Plot of PC1 and PC2 for trajectories of simulation
According to the data in Fig. 8, PC1, and PC2 were bearing 86.05 and 3.86 % of the data based on their eigenvalues, respectively.
A primary docking of known ligands with antagonist activity together with a series of decoys was performed on representative frames and the resulted dlgs were subjected for calculation of ROC values. The area under the curve for all representative frames are displayed in Table 3. The highest values of ROC were observed in frames 1638 (0.767), 2677 (0.722), and 5337 (0.775), suggesting reasonable predictive ability for these models of human dopamine D4 receptor. Since all used structures were previously evaluated for antagonist activity, the frames were therefore representing the receptor at antagonist state. Although the described frames with reasonable results of ROC values were extracted from different clusters, all of them were bearing the conserved ionic lock moiety needed for antagonist state of the receptor. In an attempt to improve the docking protocol, the procedure was repeated on the described frames using a more time consuming method (run = 100, evaluation = 25,000,000, population size = 300). As displayed in Table 3 and Fig. 9, the modified docking protocol led to a significant increase in ROC value in case of frame 1638 (0.839). Tuning of docking parameters was however futile in the case of the other two frames used in this experiment. In addition to analysis of binding energies for the ligands and receptor complexes, the binding mode of some docked structures was investigated by visual inspection. The proposed binding mode of CHEMBL332154; the selective D4 antagonist [54]; possessed the lowest docking energy, as displayed in Fig. 10. The three main residues which are crucial in the interaction of the ligand and human dopamine D4 receptor are Trp407, Asp115, and Ser122. As displayed in Fig. 10, residue Trp407 could take role in π-π and cation-π interactions with the ligand. In addition, Asp115 and Ser122 reinforced the contacts by formation of ionic bond and hydrogen bond interactions, respectively. Superposition of the best frame of D4 with the crystal structure of D3 at TM regions has revealed and RMSD value of 2.42. The superposed structures are provided in the Supplementary information section in order to avoid lengthening of the paper. All described observations demonstrated that the final optimized model is representative of D4 receptor at antagonist state.

ROC curve of frame 1638 with an improved docking protocol (run = 100, evaluation = 25,000,000, population size = 300)

Docking result for the interaction of human dopamine D4 receptor with CHEMBL332154
Conclusions
A 3D model for human dopamine D4 receptor was presented based on homology modeling, docking and molecular dynamic simulation studies. Verification of model using ramachandran plot showed that most of these residues are in favored or allowed regions of the plot. The presence of ionic lock as well as a conserved disulfide bridge in our proposed model is also additive to its advantages as a proper 3D structure. The further validation of the model by docking studies showed that this model could mimic the antagonistic state of the receptor with a reasonable ability to distinguish between ligands and decoys (ROC value = 0.839). It could be concluded that the presented 3D model of D4 receptor could donate an appropriate insight for designing new ligands in future studies.
References
Fanelli F, De Benedetti PG (2011) Update 1 of: computational modeling approaches to structure–function analysis of G protein-coupled receptors. Chem Rev 111(12):438–535
Bokoch MP, Zou Y, Rasmussen SG, Liu CW, Nygaard R, Rosenbaum DM, Fung JJ, Choi H-J, Thian FS, Kobilka TS (2010) Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature 463(7277):108–112
Rosano C, Lappano R, Santolla MF, Ponassi M, Donadini A, Maggiolini M (2012) Recent advances in the rationale design of GPER ligands. Curr Med Chem 19(36):6199–6206
Xiang L, Szebeni K, Szebeni A, Klimek V, Stockmeier CA, Karolewicz B, Kalbfleisch J, Ordway GA (2008) Dopamine receptor gene expression in human amygdaloid nuclei: elevated D4 receptor mRNA in major depression. Brain Res 1207:214–224
Beaulieu J-M, Gainetdinov RR (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 63(1):182–217
Strange B, Gartmann N, Brenninkmeyer J, Haaker J, Reif A, Kalisch R, Büchel C (2014) Dopamine receptor 4 promoter polymorphism modulates memory and neuronal responses to salience. NeuroImage 84:922–931
Furth KE, Mastwal S, Wang KH, Buonanno A, Vullhorst D (2013) Dopamine, cognitive function, and gamma oscillations: role of D4 receptors. Front Cell Neurosci 7:102. doi:10.3389/fncel.2013.00102
Enguehard-Gueiffier C, Hübner H, El Hakmaoui A, Allouchi H, Gmeiner P, Argiolas A, Melis MR, Gueiffier A (2006) 2-[(4-phenylpiperazin-1-yl) methyl] imidazo (di) azines as selective D4-ligands. Induction of penile erection by 2-[4-(2-methoxyphenyl) piperazin-1-ylmethyl] imidazo [1, 2-a] pyridine (PIP3EA), a potent and selective D4 partial agonist. J Med Chem 49(13):3938–3947
Boyfield I, Brown TH, Coldwell MC, Cooper DG, Hadley MS, Hagan JJ, Healy MA, Johns A, King RJ, Middlemiss DN (1996) Design and synthesis of 2-naphthoate esters as selective dopamine D4 antagonists. J Med Chem 39(10):1946–1948
Berry CB, Bubser M, Jones CK, Hayes JP, Wepy JA, Locuson CW, Daniels JS, Lindsley CW, Hopkins CR (2014) Discovery and characterization of ML398, a potent and selective antagonist of the D4 receptor with in vivo activity. ACS Med Chem Lett 5(9):1060–1064
Sampson D, Zhu XY, Eyunni SVK, Etukala JR, Ofori E, Bricker B, Lamango NS, Setola V, Roth BL, Ablordeppey SY (2014) Identification of a new selective dopamine D4 receptor ligand. Bioorg Med Chem 22(12):3105–3114. doi:10.1016/j.bmc.2014.04.026
Arora J, Bordeleau M, Dube L, Jarvie K, Mazzocco L, Peragine J, Tehim A, Egle I (2005) N-[(3S)-1-Benzylpyrrolidin-3-yl]-(2-thienyl)benzamides: human dopamine D4 ligands with high affinity for the 5-HT2A receptor. Bioorg Med Chem Lett 15(23):5253–5256. doi:10.1016/j.bmcl.2005.08.051
Abdelfattah MAO, Lehmann J, Abadi AH (2013) Discovery of highly potent and selective D4 ligands by interactive SAR study. Bioorg Med Chem Lett 23(18):5077–5081. doi:10.1016/j.bmcl.2013.07.033
Boström J, Böhm M, Gundertofte K, Klebe G (2003) A 3D QSAR study on a set of dopamine D4 receptor antagonists. J Chem Inf Comput Sci 43(3):1020–1027
Fleishman SJ, Unger VM, Ben-Tal N (2006) Transmembrane protein structures without X-rays. Trends Biochem Sci 31(2):106–113. doi:10.1016/j.tibs.2005.12.005
Shahlaei M, Madadkar-Sobhani A, Fassihi A, Saghaie L (2011) Exploring a model of a chemokine receptor/ligand complex in an explicit membrane environment by molecular dynamics simulation: the human CCR1 receptor. J Chem Inf Model 51(10):2717–2730
De Brevern AG (2010) 3D structural models of transmembrane proteins. In: Membrane protein structure determination. Springer, Berlin pp 387–401
Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V (2010) Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330(6007):1091–1095
Elbegdorj O, Westkaemper RB, Zhang Y (2013) A homology modeling study toward the understanding of three-dimensional structure and putative pharmacological profile of the G-protein coupled receptor GPR55. J Mol Graph Model 39:50–60
Becker O, Shacham S, Marantz Y, Noiman S (2003) Modeling the 3D structure of GPCRs: advances and application to drug discovery. Current opinion in drug discovery & development 6(3):353–361
Nowak M, Kolaczkowski M, Pawlowski M, Bojarski AJ (2006) Homology modeling of the serotonin 5-HT1A receptor using automated docking of bioactive compounds with defined geometry. J Med Chem 49(1):205–214
Platania CBM, Salomone S, Leggio GM, Drago F, Bucolo C (2012) Homology modeling of dopamine D2 and D3 receptors: molecular dynamics refinement and docking evaluation. PLoS One 7(9):e44316
Platania CBM, Leggio GM, Drago F, Salomone S, Bucolo C (2013) Regulation of intraocular pressure in mice: structural analysis of dopaminergic and serotonergic systems in response to cabergoline. Biochem Pharmacol 86(9):1347–1356
Costanzi S (2013) Modeling G protein-coupled receptors and their interactions with ligands. Curr Opin Struct Biol 23(2):185–190
Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A (2003) ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 31(13):3784–3788
Katritch V, Cherezov V, Stevens RC (2012) Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol Sci 33(1):17–27
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23(21):2947–2948. doi:10.1093/bioinformatics/btm404
Tusnady GE, Simon I (1998) Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J Mol Biol 283(2):489–506
Sonnhammer EL, von Heijne G, Krogh A (1998) A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol 6:175-182
Cserzö M, Wallin E, Simon I, von Heijne G, Elofsson A (1997) Prediction of transmembrane alpha-helices in prokaryotic membrane proteins: the dense alignment surface method. Protein Eng 10(6):673–676
Hirokawa T, Boon-Chieng S, Mitaku S (1998) SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14(4):378–379
Hofman K (1993) TMbase-A database of membrane spanning protein segments. Biol Chem Hoppe-Seyler 374:166
Käll L, Krogh A, Sonnhammer EL (2005) An HMM posterior decoder for sequence feature prediction that includes homology information. Bioinformatics 21(suppl 1):i251–i257
Rost B, Yachdav G, Liu J (2004) The predictprotein server. Nucleic Acids Res 32(suppl 2):W321–W326
Shokri A, Abedin A, Fattahi A, Kass SR (2012) J Am Chem Soc 134:10646–10650
Bernsel A, Viklund H, Hennerdal A, Elofsson A (2009) TOPCONS: consensus prediction of membrane protein topology. Nucleic Acids Res: gkp363
Šali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234(3):779–815
Eswar N, Eramian D, Webb B, Shen MY, Sali A (2008) Protein structure modeling with MODELLER. Methods Mol Biol 426:145–159. doi:10.1007/978-1-60327-058-8_8
Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9(1):40
Ko J, Lee D, Park H, Coutsias EA, Lee J, Seok C (2011) The FALC-Loop web server for protein loop modeling. Nucleic Acids Res 39(suppl 2):W210–W214
Buchan DW, Minneci F, Nugent TC, Bryson K, Jones DT (2013) Scalable web services for the PSIPRED protein analysis workbench. Nucleic Acids Res 41(W1):W349–W357
Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26(2):283–291
Schmidt TH, Kandt C (2012) LAMBADA and InflateGRO2: efficient membrane alignment and insertion of membrane proteins for molecular dynamics simulations. J Chem Inf Model 52(10):2657–2669. doi:10.1021/ci3000453
Kandt C, Ash WL, Peter Tieleman D (2007) Setting up and running molecular dynamics simulations of membrane proteins. Methods 41(4):475–488
Allen WJ, Lemkul JA, Bevan DR (2009) GridMAT-MD: a grid-based membrane analysis tool for use with molecular dynamics. J Comput Chem 30(12):1952–1958. doi:10.1002/jcc.21172
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14(1):33–38
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ (2005) GROMACS: fast, flexible, and free. J Comput Chem 26(16):1701–1718. doi:10.1002/jcc.20291
Laurie AT, Jackson RM (2005) Q-SiteFinder: an energy-based method for the prediction of protein–ligand binding sites. Bioinformatics 21(9):1908–1916
Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B (2012) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res 40(D1):D1100–D1107
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open Babel: an open chemical toolbox. J Cheminformatics 3(1):1–14
Triballeau N, Acher F, Brabet I, Pin J-P, Bertrand H-O (2005) Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J Med Chem 48(7):2534–2547
Sakhteman A, Lahtela-Kakkonen M, Poso A (2011) Studying the catechol binding cavity in comparative models of human dopamine D2 receptor. J Mol Graph Model 29(5):685–692. doi:10.1016/j.jmgm.2010.11.012
Vogel R, Mahalingam M, Lüdeke S, Huber T, Siebert F, Sakmar TP (2008) Functional role of the “ionic lock”—an interhelical hydrogen-bond network in family A heptahelical receptors. J Mol Biol 380(4):648–655
Kesten SR, Heffner TG, Johnson SJ, Pugsley TA, Wright JL, Wise LD (1999) Design, synthesis, and evaluation of chromen-2-ones as potent and selective human dopamine D4 antagonists. J Med Chem 42(18):3718–3725
Acknowledgments
The support from the Research Council at Shiraz University of Medical Sciences is acknowledged. The authors would like to thank Nazanin Bagherzadeh for her kind contribution in language editing of the manuscript. This work is a PharmD dissertation report, performed by Minasadat Khoddami, student of pharmacy at Shahid Sadoughi University of Medical Sciences.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Khoddami, M., Nadri, H., Moradi, A. et al. Homology modeling, molecular dynamic simulation, and docking based binding site analysis of human dopamine (D4) receptor. J Mol Model 21, 36 (2015). https://doi.org/10.1007/s00894-015-2579-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-015-2579-3