
Abstract
Constitutive activation of extracellular signal-regulated kinase (ERK)1/2 pathway, that is activated by various stimuli including growth factors and oncogenic driver mutations, is observed in various cancers. However, the difference of the activated levels of the pathway is still unclear in clinical significances. The aim of this study was to investigate the effect of different ERK1/2 pathway activation, assessed by the expression levels of phosphorylated (p) ERK1/2, on the prognosis of advanced lung adenocarcinoma patients. Paraffin-embedded lung biopsy samples were obtained from 85 lung adenocarcinoma patients. Correlation between pERK1/2 expression levels that were assessed by immunohistochemistry (IHC) analysis and oncogenic driver mutation status, clinicopathological factors, outcome from standard anticancer therapies, and prognosis was investigated. Varying levels of pERK1/2 expression were observed in 68 (80.0 %) patients. The overall survival was significantly reduced in patients with higher pERK1/2 expression in comparison to those with lower expression levels (P = 0.03). In particular, higher pERK1/2 expression levels correlated with worse performance status and worse clinical outcome. Thus, the IHC analysis of pERK1/2 expression levels may predict patient prognosis in advanced lung adenocarcinoma. Inhibition of ERK1/2 pathway activated by various signals may improve the effects of standard chemotherapies and the clinical condition of patients with advanced cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lung cancer is the most frequent cause of cancer-related deaths worldwide, accounting for more than 1 million deaths annually [1]. Traditionally, treatment strategy of lung cancer has been based on histological considerations, with tumors being classified into two major histological types: small cell lung cancers (SCLCs) and non-small cell lung cancers (NSCLCs) [2]. NSCLCs are classified to adenocarcinoma, squamous cell carcinoma, and large cell carcinoma. NSCLC represents approximately 80 % of all lung malignancies and is associated with a poor 5-year survival rate of around 15 %. Multiple genetic and epigenetic events were reported to be involved in the development of NSCLC [3], with a number of so-called driver mutations identified as potential therapeutic targets in particular in lung adenocarcinoma [4].
Oncogenic driver mutations cause constitutive activation of signaling pathways, which lead to uncontrolled cell growth and proliferation. Signaling via the mitogen-activated protein kinase (MAPK) pathway, constituted of several intracellular signaling cascades including the rat sarcoma viral oncogene homolog (RAS)/raf proto-oncogene serine-threonine kinase (RAF)/MAPK extracellular signal-regulated kinase (ERK) (MEK)/ERK1/2, and the Ras/phosphatidylinositol 3-kinase (PI3K)/phosphatase and tensin homolog (PTEN)/v-akt murine thymoma viral oncogene homolog (AKT)/mechanistic target of rapamycin (mTOR) pathways, is a serious of carefully orchestrated events, generally starting with a stimulus on the cell surface which is transferred via sequential phosphorylation events to regulatory proteins controlling gene transcription within the nucleus. Regulation of these pathways is mediated by a series of kinases, phosphatases, and exchange proteins [5]. ERK1 represents the p44 MAPK3 protein and ERK2 represents p42 MAPK1 protein. The two homologous proteins are involved in the same pathway. The RAS/RAF/MEK/ERK1/2 pathway is activated through the activation of cell surface receptor tyrosine kinases such as epidermal growth factor receptor (EGFR), platelet derived growth factor receptor (PDGFR), vascular endothelial growth factor receptor (VEGFR) and G-protein coupled receptors (GPCRs) by growth factors, mitogens and cytokines [6–8]. Activated ERK1/2 is finally phosphorylated on the TxY motif (Thr202/Tyr204) within its activation loop, completing the cascade [8–11]. Chappell et al. [5] presented an overview of the effects of mutations and activation of these signaling pathways and reported that dysregulated expression of growth factor receptors by increased expression, genetic translocations, or genomic amplifications may lead to activation of the RAS/RAF/MEK/ERK1/2 and RAS/PI3K/PTEN/AKT/mTOR pathways. Alternatively, chromosomal translocations can occur in non-receptor kinases and other genes, thereby resulting in activation of these pathways [5].
EGFR, Kirsten rat sarcoma viral oncogene homolog (KRAS), and v-raf murine sarcoma viral oncogene homolog B (BRAF) mutations have all been demonstrated to cause constitutive activation of their downstream effectors, resulting in increased phosphorylation and activation of ERK1/2, causing enhanced signaling to the cytosolic and nuclear effector proteins and increased cellular proliferation, differentiation, growth, and survival [12, 13].
In NSCLC, particular in lung adenocarcinoma, EGFR mutation analysis is the best biomarker to predict the therapeutic relevance of EGFR-tyrosine kinase inhibitors (TKIs) gefitinib and erlotinib. The clinical and biological data concerning EGFR have been extensively reviewed elsewhere [14, 15]. Echinoderm microtubule-associated protein-like 4 (EML4)–anaplastic lymphoma kinase (ALK) fusion genes are also important in lung adenocarcinoma as driver mutations that influence the effectiveness of molecular targeted therapy with crizotinib [16, 17] followed by EGFR-TKIs for patients with EGFR mutations.
In lung cancer, KRAS mutations were the first driver mutations to be discovered [18]. KRAS mutations have been reported to correlate with poor patient prognosis. Although EGFR and KRAS are part of the same signaling pathway, the role of KRAS mutations in lung cancer remains to be elucidated [12, 13, 19, 20]. KRAS mutations are not targets for chemical therapeutic agents but can be treated with combined MEK inhibitors and PI3K or AKT inhibitors [21, 22]. BRAF mutations cause constitutive activation of the pathways downstream of ERK1/2, and occur more frequently in smokers [18, 23].
In this study, the immunohistochemical nuclear expression levels of pERK1/2 as a result of transcriptional activity were investigated in advanced lung adenocarcinoma patients who had not received systemic anticancer treatment. Several previous studies have reported that active EGFR signaling causes phosphorylation of ERK1/2, and decreased expression of pERK1/2 is regarded as a marker of successful inhibition of the MAPK signaling pathway in a variety of cancers [24, 25]. However, in NSCLC including lung adenocarcinoma, potential variations in pERK1/2 expression levels prior to anticancer treatment have never been reported. Hence, the aim of this study was to clarify the correlation between expression levels of pERK1/2 and prognosis in patients with advanced lung adenocarcinoma, in relation with the gene mutation status of EGFR, KRAS, and BRAF, and ALK rearrangements. Furthermore, we investigated the correlation between pERK1/2 expression levels and the standard chemotherapeutic response in advanced lung adenocarcinoma without molecular therapeutic targets. The prognosis of advanced cancer patients is thought to be dependent on the response to anticancer treatments. Molecular targeted therapies have been improved and are expected to be more effective; however, many patients lacking target molecules are subjected to standard chemotherapy and the effectiveness are not complete. Therefore, we investigated whether the variation in the levels of pERK1/2 expression resulting from differential ERK1/2 pathway activation in advanced lung adenocarcinomas, can serve as a prognostic marker for standard anticancer treatments and become a therapeutic target.
Materials and methods
Patients and tumor tissue samples
Formalin-fixed and paraffin-embedded lung biopsy specimens were obtained from 172 patients with lung carcinomas who underwent tracheobronchoscopic tumor biopsy (TBTB) at the Department of Respiratory Medicine, Nihon University Itabashi Hospital between January 2009 and December 2010. One hundred and seventeen patients were diagnosed with adenocarcinoma. Immunohistochemistry (IHC) analysis showed that they were positive for the lung adenocarcinoma markers; thyroid transcription factor-1 (TTF-1) and/or cytokeratin (CK) 7, and negative for CK20, a metastatic carcinoma marker [26], squamous cell carcinoma suggesting antibody, p63 [27], CK5/6 and CK14 [28], and neuroendocrine markers CD56 and synaptophysin [29]. Out of the 117 patients, 85 with complete clinical information who had not received any anticancer treatments before the TBTB were included in this retrospective study. The study was approved by the institutional review board of Nihon University Itabashi Hospital, Tokyo, Japan (RK-121109-9). Eighty-five formalin-fixed and paraffin-embedded (FFPE) TBTB specimens were obtained in this study.
Immunohistochemistry
We prepared 4 µm FFPE sections for IHC. Phosphorylated ERK1/2 and non-phosphorylated ERK1/2 were assessed by IHC analysis using a rabbit monoclonal anti-phospho-ERK1/2 antibody (Thr202/Tyr204, D13.14.4E XP™; Cell Signaling Technology Inc., MA, USA) and a mouse monoclonal anti-ERK1/2 antibody (clone MK12; Millipore, CA, USA). The slides were deparaffinized, and antigen retrieval was performed by heating the slides in citrate buffer (pH 6.0) for 15 min at 121 °C followed by cooling down to room temperature (RT). After washing with phosphate-buffered saline (PBS), endogenous peroxidase was blocked with 3 % hydrogen peroxide for 5 min, and the slides were washed with PBS and incubated with the primary antibodies overnight at 4 °C. Next, after washing with PBS, the slides were incubated with the polymer secondary antibody (Simple Stain Max PO Multi; Nichirei Bioscience, Kyoto, Japan) for 30 min at RT. The slides were again washed with PBS, and subsequently incubated with 3,3-diaminobenzidine (DAB) for 10 min. Counterstaining with hematoxylin was performed for 30 s in order to visualize the nuclei.
The nuclear pERK1/2 expression levels were assessed on the modified IHC Allred score by trained histopathologists. The intensity score (IHC-IS) was evaluated as 0, 1+, 2+, or 3+, and the proportion score (IHC-PS) was evaluated as 0, 1, 2, 3, or 4 (0, <10, 10–50, 51–80, and >80 % of positively stained tumor nuclei, respectively). Although the original Allred score was sum of IHC-PS and IHC-IS, the present total IHC scores were calculated as multiply of IHC-PS and IHC-IS, for clarify the significance of scores by reference to H score (multiply of the percentage of positive staining cells and the intensity score) [30], and were classified as negative (0), low (1–2), moderate (3–4), or high (6–12).
EGFR, KRAS and BRAF gene mutation assays from FFPE biopsy specimens
Target tumor cells were microdissected from FFPE TBTB sections using the PALM® MicroBeam III-N (Carl Zeiss Microscopy, Munich, Germany) as previously described [31, 32]. In brief, 8-µm-thick FFPE sections were mounted on membrane film-coated glass slides. Following dewaxing with xylene and ethanol, the sections were stained with toluidine blue, and air-dried. The targeted tumor cells were then microdissected and retrieved precisely into an Eppendorf lid with mineral oil. Samples were lysed in 200 μl of DNA lysis buffer that contained 2 % sodium dodecyl sulfate, 0.1 mM ethylenediaminetetraacetic acid (EDTA), and 10 mM Tris–HCl, and incubated overnight at 55 °C with proteinase K. DNA was purified with 100 μl of buffer-saturated phenol (pH 8.0) and 100 μl of chloroform–isoamyl (24:1) alcohol. Following centrifugation, the upper aqueous layers were transferred into fresh tubes. Ammonium acetate pH 8.0 (100 μl) and ethanol (400 μl) were added, and the samples were subsequently frozen at −20 °C for 30 min. The DNA pellets were collected by centrifugation for 30 min at 15,000 rpm, air-dried, and dissolved in 10 μl Tris–EDTA buffer. The DNA samples were stored at 4 °C.
EGFR, KRAS and BRAF mutations were detected by melting temperature (TM) analysis of polymerase chain reaction (PCR) products amplified with quenching probes (QP) using the fully automated genotyping system i-densy ™ 5320 (Arkray Inc., Kyoto, Japan), and this techniques have been called as QP methods [33–35]. In the present study, hot spots of EGFR (exon 18–21), KRAS (exon 2–3), and BRAF (exon 15) were analyzed using the specific QPs and PCR primers (NIPPON STEEL & SUMIKIN Eco-Tech Co., Tokyo, Japan). The primer sequences used for amplifications and the QP sequences were as follows: EGFR exon 19. Forward primer: 5′-tctctctgtcatagggactc-3′, Reverse primer: 5′-gaaactcacatcgaggatttc-3′, QP: 5′-cccgtcgctatcaaggaattaagagaagc-3′; EGFR exon 20. Forward primer: 5′-tccaggaagcctacgtgatggccag-3′, Reverse primer: 5′-ccaatattgtctttgtgttcccggacatagtc-3′, QP: 5′-tgagctgcatgatgaggtgcac-3′; EGFR exon 21. Forward primer: 5′-aggaacgtactggtgaaaacaccgc-3′, Reverse primer: 5′-gcctccttctgcatggtattctttctc-3′, QP: 5′-ttggcccgcccaaaatc -3′; KRAS exon 2–3. Forward primer: 5′-aaggcctgctgaaaatgactg-3′, Reverse primer: 5′-ggtcctgcaccagtaatatgca -3′, QP: 5′-ctcttgcctacgccaccagctccaact -3′; BRAF exon 15. Forward primer: 5′-tgcttgctctgataggaaaatgagatctac-3′, Reverse primer: 5′-aaactgatgggacccactccat-3′, QP: 5′-gctacagagaaatctc-3′. Purified DNA samples and the target primers and probes were added to commercial reagent packs including PCR master mix (Arkray, Inc.). Using the i-densy™ 5320, PCR was performed with an initial denaturation for 1 min at 95 °C before 50 cycles of denaturation at 95 °C for 1 s and annealing at 60 °C for 15 s. After the PCR was complete, TM analyses were automatically performed. Mutations were detected as peaks of TM differing from the wild type [33]. PCR products were purified using Microcon 100 (Takara Bio Inc., Shiga, Japan), and sequencing analyses was performed using the ABI 310 genetic analyzer (Thermo Fisher Scientific Inc., Waltham, MA, USA) with the same PCR primers and the Big Dye Terminator v1.1 Cycle Sequencing Kit (Thermo Fisher Scientific Inc.) according to the manufacturer’s instructions.
ALK rearrangement detection
Anaplastic lymphoma kinase (ALK) rearrangements were detected with IHC using ALK Detection Kit (Nichirei Bioscience, Tokyo, Japan), which is based on the intercalated antibody-enhanced polymer (iAEP) method [36] and includes the 5A4 clone as the anti-ALK primary antibody. Stronger cytoplasmic staining in tumor cells in comparison to negative control cells was evaluated as positive based on the iScore [36]. Total RNA was extracted from microdissected FFPE samples staining positive for ALK upon IHC analysis, as previously described [31, 32]. ALK fusion transcripts were confirmed using multiplex reverse transcription (RT)-PCR methods [37] and sequenced using the ABI 310 genetic analyzer (Thermo Fisher Scientific Inc.) and the Big Dye Terminator v1.1 Cycle Sequencing Kit (Thermo Fisher Scientific, Inc.) according to the manufacturer’s instructions (Table 1).
Statistical analyses
Significant differences in the pERK1/2 IHC expression scores were analyzed using the Mann–Whitney U test and Pearson’s Chi squared test. Relative risk of anti-tumor drug resistance rate was analyzed for every pERK1/2 expression level. Prognostic analyses were performed using Kaplan–Meier survival curves, log-rank tests, and Cox regression models. All analyses were performed using SPSS® Statistics version 20.0 (IBM Japan, Tokyo, Japan).
Results
Differences of immunohistochemical pERK1/2 expression levels in lung adenocarcinomas
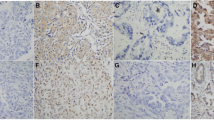
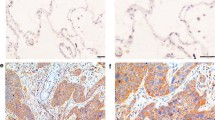
Phosphorylated ERK1/2 was detected in 68 (80.0 %) of the 85 patients (Table 2). IHC analysis showed nuclear and cytoplasmic expression of pERK1/2 in tumor cells. In the present study, the only nuclear staining was evaluated as an indication of ERK1/2 transcriptional activity. Variable pERK1/2 IHC expression levels among patients are shown in Fig. 1b–d. Phosphorylated ERK1/2 expression levels were low in 35 (41.2 %) (Fig. 1c), and high in 33 patients (38.8 %) (Fig. 1d). Non-phosphorylated ERK1/2 expression was observed in all cases (see Fig. 1g) and no difference in expression levels were detected among samples.

Immunohistochemical staining of pERK1/2 and ERK1/2 protein expression. Positive control of a pERK1/2 and e ERK1/2 expressed in blood vessels (arrows). In lung adenocarcinoma, pERK1/2 is expressed in both the tumor cytoplasm and nuclei, but only nucleic staining was evaluated: b negative, c low, and d high nuclear pERK1/2 expression. f Negative control of ERK1/2. g Non-phosphorylated ERK1/2 is expressed in the tumor cytoplasm and nuclei of all present samples regardless of the phosphorylation status. Each scale bar represents 20 μm
The correlations between pERK1/2 expression levels and clinicopathological characteristics are shown in Table 3. Age, gender, smoking status, and histopathological differentiation did not significantly correlate with patient survival or pERK1/2 expression levels. Stage IV patients appeared to have worse outcomes than other patients (P = 0.07), but none of the clinical stages significantly correlated with pERK1/2 expression. No significant difference in pERK1/2 expression levels was observed between patients who survived for more than 3 years after diagnosis and those who did not. However, performance status (PS)4 patients evaluated by the Eastern Cooperative Oncology Group (ECOG) performance status [38] had significantly worse outcomes than PS0 patients (median overall survival [OS] times were 16.5 and 3.5 months, respectively; P = 0.0006), and PS3 patients had worse outcomes than PS0 patients (median OS times were 16.5 and 6.0 months, respectively; P = 0.004). pERK1/2 expression levels in PS4 patients were significantly higher than in PS0 patients.
pERK1/2 expression level and gene mutation status
Among the patients, EGFR mutations were detected in 16 (18.8 %), KRAS mutations in 11 (12.9 %), BRAF mutations in 2 (2.4 %), and EML4-ALK fusion in 1 (1.2 %). The rates of occurrence of the EGFR, KRAS, and BRAF mutations, and the ALK rearrangements according to the pERK1/2 expression levels are shown in Fig. 2a. The patients with EGFR gene mutations were in the lower pERK1/2 expression groups, and the patients with KRAS mutations were in the higher pERK1/2 expression groups (P = 0.002, Pearson’s Chi squared test). In these patients, a BRAF mutation was detected in only two patients, and only one patient had an ALK rearrangement. Among the BRAF mutated patients, pERK1/2 expression levels appeared to be high and no pERK expression was observed in ALK-positive patient. pERK1/2 expression levels were significantly higher in patients with KRAS mutations than in those with EGFR mutations (P = 0.0005), and in patients with BRAF mutations than in EGFR mutations (P = 0.044) (Fig. 2b).

Occurrence rates of EGFR mutations (EGFRm, gray bar), KRAS mutations (KRASm, black bar), BRAF mutations (BRAFm, doted bar), and EML4–ALK gene fusion (white bar) in patients for each pERK1/2 expression level are shown. The patients with EGFR gene mutations were in the lower pERK1/2 expression groups, and the patients with KRAS mutations were in the higher pERK1/2 expression groups (P = 0.002, Pearson’s Chi squared test)
Survival analysis in clinicopathological factors, gene mutations, and pERK1/2 expression levels
The results of survival analyses performed using a Cox proportional hazards model are shown in Table 4. Clinicopathological factors, age, gender, smoking status, and clinical stage were not significantly correlated with patient outcomes. Patients with EGFR mutations (KRAS wild type) appeared to have better outcomes than both EGFR and KRAS wild type patients. Patients with KRAS mutations appeared to have worse outcomes than patients with EGFR mutations (P = 0.07, log-rank, Fig. 3a). Outcomes were significantly worse in patients with high pERK1/2 expression levels in comparison to those with negative or low pERK1/2 expression levels (P = 0.02, Table 4). The median overall survival times were 22.2, 18.1, and 11.6 months in patients with negative, low, moderate/high pERK1/2 expression, respectively. Moderate or high pERK1/2 expression was associated with a significantly worse overall survival than negative pERK1/2 expression (P = 0.03, log-rank, Fig. 3b).

Prognostic analyses of lung adenocarcinoma patients classified according to EGFR and KRAS mutation status (a) and pERK1/2 expression levels (b). The overall survival (OS) of patients was correlated with the pERK1/2 expression level. In particular, overall survival (OS) of patients with moderate/high pERK1/2 expression was significantly shorter than that of patients with negative pERK1/2 expression (P = 0.03, log-rank test)
pERK1/2 expression level and therapeutic outcome
Although substantial advances in molecularly targeted therapies have been made in recent years, platinum-based chemopreventive agents are still administered to many patients. Since patient prognosis primarily depends on the therapeutic effects, we next investigated the correlation between pERK expression levels and the outcome from standard chemotherapy for advanced lung adenocarcinoma at the first line treatments, including carboplatinum (CBDCA) + pemetrexed, CBDCA + paclitaxel (TXL), cisplatinum (CDDP) + vinorelbin (VNR) + radiotherapy (RT).
The correlation between pERK1/2 expression levels and the responsiveness to standard chemotherapy for advanced lung adenocarcinoma (except for EGFR-TKI treatment) is shown in Table 5. Patients with a lower pERK1/2 expression had a better response rate to standard chemotherapy. In patients with negative, low, moderate, and high pERK1/2 expression, therapeutic response rates were 85.7, 81.3, 66.7, and 50.0 %, respectively. The relative risk for the anticancer drug resistance rate in patients with low, moderate, and high pERK1/2 expression were 1.5 folds, 2.5 folds, and 3.5 folds compared with negative pERK1/2 expression group. Median survival time in the patients with high pERK1/2 expression was significantly shorter than in those with lower and negative pERK1/2 expression (P = 0.040). Negative expression of pERK1/2 appeared to be more common in patients with younger age; however, this observation was not significant. PS of patients appeared to be better in the negative pERK1/2 expression group; however, this difference was not significant.
The patients with progressive disease (PD) subjected to standard chemotherapies appeared to have higher IHC pERK1/2 expression levels than those with partial response (PR), and even higher pERK1/2 expression was observed in the patients not subjected to any anticancer treatment (best supportive care; BSC, P = 0.02) (Fig. 4a). The results of pERK1/2 expression levels for each PS grade are shown in Fig. 4b. pERK1/2 expression levels were significantly higher in PS4 patients than in PS0 patients (P = 0.04), and in PS1 patients (P = 0.007). Lower PS patients (PS0 and PS1) had lower pERK1/2 expression levels than higher PS patients (PS3 and PS4, P = 0.007).

The correlation between pERK1/2 IHC score and the response to standard chemotherapeutics of advanced lung adenocarcinoma (a), and the ECOG performance status [38] (b) are shown. a The patients with partial responses (PR) tended to have lower pERK1/2 IHC expression levels than those with progressive disease (PD), and significantly lower pERK1/2 than those not subjected to any anticancer treatment (best supportive care: BSC, P = 0.02). B: pERK1/2 expression level was significantly higher in PS4 patients than in PS0 patients (P = 0.04), and in PS1 patients (P = 0.007). Lower PS patients (PS0 and PS1) had lower pERK1/2 expression levels than higher PS patients (PS3 and PS4, P = 0.007)
Discussion
Genetic alterations that result in persistent activation of the RAS/RAF/MEK/ERK1/2 signaling pathway are common in various types of cancers [39]. This hyperactivation results are caused in stepwise phosphorylation of the downstream targets of the mutated gene, and consequently, in constitutive activation of growth signal transduction [39]. ERK1/2 activation is important for normal epidermal cell growth, and a correlation between ERK1/2 signaling and tumor malignancy has been suggested [40]. ERK1/2, a downstream target of EGFR signaling, is phosphorylated following growth factor stimulation, and reduction of pERK1/2 expression levels after effective anticancer treatment has been reported in a number of cancers. However, to our knowledge, pERK1/2 expression has not been previously reported in NSCLC patients who had not received anticancer treatments. In this study, we found varied pERK1/2 expression levels in untreated patients with advanced lung adenocarcinoma.
In the present study, as the major target gene mutations of RAS/RAF/MEK/ERK1/2 signaling pathway, EGFR, KRAS, and BRAF mutations were investigated. Although the patient series antedated the widespread use of EGFR-TKIs, we found that the patients with EGFR gene mutations had better outcomes than the patients with KRAS gene mutations. The nuclear expression levels of pERK1/2 in the adenocarcinoma cells differed significantly between the patients with EGFR mutations and the patients with KRAS mutations. Although patients with BRAF mutations were rare in the present series, their outcomes were worse (data not shown) and their pERK1/2 expression levels were high, more comparable to the patients with KRAS mutations than to those with EGFR mutations. Only one patient in the current patient series was identified to have an EML4-ALK fusion with no detectable pERK1/2 expression. Further investigation of pERK1/2 expression in patients with ALK rearrangement is required.
These results suggest that lung adenocarcinomas with downstream gene mutations on the EGFR signaling pathway might have higher signaling activity than those with EGFR gene mutations. Kubota et al. [41] reported that oncogenic RAS efficiently activates the ERK1/2 pathway, both by activating RAF and by inhibiting MEK sumoylation, which is an important mechanism for modulation of cellular function. In fact, although ERK1/2 expression levels were equal in almost all of the patients in the present study, the expression levels of pERK1/2 were significantly higher in patients with KRAS gene mutations than in patients with EGFR gene mutations. Although inhibition of the ERK1/2 pathway was associated with a G1 cell cycle arrest in cells with RAS mutations [42], apoptosis was not remarkably induced in TKI sensitive lung adenocarcinoma cells [43]. These results showed that EGFR mutant lung adenocarcinomas might have another driving pathway for tumor progression other than the ERK1/2 pathway.
Lung adenocarcinoma is known for its diverse morphology constituted of adenocarcinoma in situ, microinvasive adenocarcinoma, and invasive adenocarcinoma subclassified to lepidic, acinar, papillary, solid, mucinous, colloid, fetal, micropapillary, etc. These morphological assessments are quite important for predicting the prognosis after surgery for early stage lung adenocarcinoma. In this study, as advanced lung adenocarcinomas were investigated using TBTB samples, the correlation between pERK1/2 expression levels and the detailed morphological pattern was not clear. Further studies have to be performed to establish a correlation between pERK1/2 expression and the morphologies of early stage lung adenocarcinoma including the prognosis after operation. Correlation with tumor invasion is important in early stage cancer prognosis as recurrence and metastasis. However, in the present invasive and higher clinical stage patients, pERK1/2 expression levels were quite different from negative to high. In patients with inoperable advanced lung adenocarcinoma, a prediction for the prognosis after anticancer treatment was required. In this study, the correlation between pERK1/2 expression and the response to standard chemotherapies was investigated. In patients with higher pERK1/2 expression, the standard chemotherapeutic response was worse and the outcome was poorer in comparison to other patients. In addition, pERK1/2 expression levels were higher in patients who were not subjected to any standard treatment for advanced lung adenocarcinoma (only best supportive care), which correlated with their poor PS and worse prognosis. These results suggested that pERK1/2 expression levels correlated with prefer to therapeutic outcome than tumor invasion. Furthermore, abnormal ERK1/2 activation induced by multiple cell signaling pathways, as demonstrated by high IHC expression levels of pERK1/2, may correlate not only with tumor progression and survival but also with worsened clinical conditions of patients. Therefore, tumor pERK1/2 overexpression in the TBTB sections of advanced lung adenocarcinoma patients could serve as a potential prognostic factor despite the variation in driver mutations. The correlation between pERK1/2 expression levels and different therapeutic regimens is required to analysis. The mechanism of therapeutic resistance in lung adenocarcinoma with higher pERK1/2 expression level, such as possible decreased potency of the anticancer drug due to increased cell growth and migration, problems with drug transport, or other reasons should be investigated.
Control or inhibition of ERK1/2 phosphorylation may represent a potential novel therapeutic target in the treatment of patients with refractory lung adenocarcinoma, including patients with KRAS mutations and those without indications for any anticancer treatments. MEK inhibitors such as selumetinib (AZD6244; AstraZeneca, Wilmington, DE, USA) have been reported in clinical trials to have promising results as a targeted therapy for patients with KRAS-mutation-positive NSCLC [44–46]. In other types of NSCLC, although the results of various exhaustive genetic analyses using next-generation sequencing techniques have been reported [47–49], no other gene mutations that could serve as therapeutic targets other than EGFR mutations and ALK rearrangements have been found. The recognized array of less common oncogenic alterations in lung adenocarcinoma, including those in the ROS1, RET, BRAF, and ERBB2 genes, is growing rapidly [47]. The therapeutic implications of these findings are, in many cases, still under investigation [47]. Genotypes are quite varied and complicated, and the correspondence of one molecular targeted therapy to one oncogene mutation may be limited. Therefore, when chemotherapies are required for advanced NSCLC patients including lung adenocarcinoma patients, and to predict the response to platinum-based standard chemotherapies, pERK1/2 evaluation by IHC may be useful. To improve the response rates to MEK inhibitors, pERK1/2 evaluation by IHC may also be useful for selecting the most appropriate subset of lung adenocarcinoma patients, despite their actual genotypes. Moreover, the development of treatments for advanced lung adenocarcinoma resistant to other anticancer treatment is anticipated. Furthermore, MEK inhibitors may be used as an adjuvant treatment for patients with poor outcome from standard anticancer treatment and with high pERK1/2 expression levels, and as a treatment to improve the worsened clinical conditions of the patients who have not received any anticancer treatment because of their worse PS.
Conclusions
Tumor nucleic pERK1/2 expression levels differed significantly in patients with advanced lung adenocarcinoma. These were higher not only in the patients with KRAS gene mutations but also in the patients who had worse outcome from standard chemotherapies and who had not received anticancer treatments because of their worse PS. Immunohistochemical evaluation of pERK1/2 expression levels may be useful for predicting chemotherapeutic outcome in advanced lung adenocarcinoma and in selecting patients who are most likely to benefit from treatment with MEK inhibitors.
References
American Cancer Society: Cancer Facts and Figures 2007 (2007) American Cancer Society, Atlanta
Hirsch FR, Corrin B, Colby TV (2004) World Health Organization classification of tumours. In: Travis WB, Brambilla A, Muller-Hermelinck HK, Harris CC (eds) Pathology and genetics of tumours of the lung, pleura, thymus and heart. IARC Press Lyon, France, p 10
Sato M, Shames DS, Gazdar AF, Minna JD (2007) A translational view of the molecular pathogenesis of lung cancer. J Thorac Oncol 2:327–343
Pao W, Girard N (2011) New driver mutations in non-small-cell lung cancer. Lancet Oncol 12:175–180
Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA, Bäsecke J, Stivala F, Donia M, Fagone P, Malaponte G, Mazzarino MC, Nicoletti F, Libra M, Maksimovic-Ivanic D, Mijatovic S, Montalto G, Cervello M, Laidler P, Milella M, Tafuri A, Bonati A, Evangelisti C, Cocco L, Martelli AM, McCubrey JA (2011) Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget 2:135–164
Geest CR, Coffer PJ (2009) MAPK signaling pathways in the regulation of hematopoiesis. J Leukoc Biol 86:237–250
Mandal R, Raab M, Matthess Y, Becker S, Knecht R, Strebhardt K (2014) pERK 1/2 inhibit Caspase-8 induced apoptosis in cancer cells by phosphorylating it in a cell cycle specific manner. Mol Oncol 8:232–249
Rudd CE (2005) MAPK p38: alternative and nonstressful in T cells. Nat Immunol 6:368–370
Cargnello M, Roux PP (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 75:50–83
Zhao Y, Adjei AA (2014) The clinical development of MEK inhibitors. Nat Rev Clin Oncol 11:385–400
Roberts PJ, Der CJ (2007) Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26:3291–3310
Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, Lorimer I, Zhang T, Liu N, Daneshmand M, Marrano P, da Cunha Santos G, Lagarde A, Richardson F, Seymour L, Whitehead M, Ding K, Pater J, Shepherd FA (2005) Erlotinib in lung cancer—molecular and clinical predictors of outcome. N Engl J Med 353:133–144
Metro G, Chiari R, Bennati C, Cenci M, Ricciuti B, Puma F, Flacco A, Rebonato A, Giannarelli D, Ludovini V, Bellezza G, Ferolla P, Minotti V, Crinò L (2014) Clinical outcome with platinum-based chemotherapy in patients with advanced nonsquamous EGFR wild-type non-small-cell lung cancer segregated according to KRAS mutation status. Clin Lung Cancer 15:86–92
Riely GJ, Politi KA, Miller VA, Pao W (2006) Update on epidermal growth factor receptor mutations in non-small cell lung cancer. Clin Cancer Res 12:7232–7241
Lindeman NI, Cagle PT, Beasley MB, Chitale DA, Dacic S, Giaccone G, Jenkins RB, Kwiatkowski DJ, Saldivar JS, Squire J, Thunnissen E, Ladanyi M (2013) College of American Pathologists International Association for the Study of Lung Cancer and Association for Molecular Pathology. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Mol Diagn 15:415–453
Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, Aburatani H, Niki T, Sohara Y, Sugiyama Y, Mano H (2007) Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448:561–566
Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Jänne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, Fidias P, Stubbs H, Engelman JA, Sequist LV, Tan W, Gandhi L, Mino-Kenudson M, Wei GC, Shreeve SM, Ratain MJ, Settleman J, Christensen JG, Haber DA, Wilner K, Salgia R, Shapiro GI, Clark JW, Iafrate AJ (2010) Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 363:1693–1703
Thunnissen E, van der Oord K, den Bakker M (2014) Prognostic and predictive biomarkers in lung cancer. A review. Virchows Arch 464:347–358
Sonobe M, Kobayashi M, Ishikawa M, Kikuchi R, Nakayama E, Takahashi T, Menju T, Takenaka K, Miyahara R, Huang CL, Okubo K, Bando T, Date H (2012) Impact of KRAS and EGFR gene mutations on recurrence and survival in patients with surgically resected lung adenocarcinomas. Ann Surg Oncol 19:S347–S354
Johnson ML, Sima CS, Chaft S, Paik PK, Pao W, Kris MG, Ladanyi M, Riely GJ (2013) Association of KRAS and EGFR mutations with survival in patients with advanced lung adenocarcinomas. Cancer 119:356–362
Rosell R, Bivona TG, Karachaliou N (2013) Genetics and biomarkers in personalization of lung cancer treatment. Lancet 382:720–731
Yang JY, Chang CJ, Xia W, Wang Y, Wong KK, Engelman JA, Du Y, Andreeff M, Hortobagyi GN, Hung MC (2010) Activation of FOXO3a is sufficient to reverse mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor chemoresistance in human cancer. Cancer Res 70:4709–4718
Cardarella S, Ogino A, Nishino M, Butaney M, Shen J, Lydon C, Yeap BY, Sholl LM, Johnson BE, Jänne PA (2013) Clinical, pathologic, and biologic features associated with BRAF mutations in non-small cell lung cancer. Clin Cancer Res 19:4532–4540
Ventura AP, Radhakrishnan S, Green A, Rajaram SK, Allen AN, O’Briant K, Schummer M, Karlan B, Urban N, Tewari M, Drescher C, Knudsen BS (2010) Activation of the MEK-S6 pathway in high-grade ovarian cancers. Appl Immunohistochem Mol Morphol 18:499–508
Shin DM, Zhang H, Saba NF, Chen AY, Nannapaneni S, Amin AR, Müller S, Lewis M, Sica G, Kono S, Brandes JC, Grist WJ, Moreno-Williams R, Beitler JJ, Thomas SM, Chen Z, Shin HJ, Grandis JR, Khuri FR, Chen ZG (2013) Chemoprevention of head and neck cancer by simultaneous blocking of epidermal growth factor receptor and cyclooxygenase-2 signaling pathways: preclinical and clinical studies. Clin Cancer Res 19:1244–1256
Linder S, Olofsson MH, Herrmann R, Ulukaya E (2010) Utilization of cytokeratin-based biomarkers for pharmacodynamic studies. Expert Rev Mol Diagn 10:353–359
Koster MI, Kim S, Mills AA, DeMayo FJ, Roop DR (2004) p63 is the molecular switch for initiation of an epithelial stratification program. Genes Dev 18:126–131
Chu PG, Weiss LM (2001) Keratin expression in human tissues and neoplasms. Histopathology 40:403–439
Zheng G, Ettinger DS, Maleki Z (2013) Utility of the quantitative Ki-67 proliferation index and CD56 together in the cytologic diagnosis of small cell lung carcinoma and other lung neuroendocrine tumors. Acta Cytol 57:281–290
Ishibashi H, Suzuki T, Suzuki S, Moriya T, Kaneko C, Takizawa T, Sunamori M, Handa M, Kondo T, Sasano H (2003) Sex steroid hormone receptors in human thymoma. J Clin Endocrinol Metab 88:2309–2317
Nakanishi Y, Shimizu T, Tsujino I, Obana Y, Seki T, Fuchinoue F, Ohni S, Oinuma T, Kusumi Y, Yamada T, Takahashi N, Hashimoto S, Nemoto N (2013) Semi-nested real-time reverse transcription polymerase chain reaction methods for the successful quantitation of cytokeratin mRNA expression levels for the subtyping of non-small-cell-lung carcinoma using paraffin-embedded and microdissected lung biopsy specimens. Acta Histochem Cytochem 46:85–96
Mizutani G, Nakanishi Y, Watanabe N, Honma T, Obana Y, Seki T, Ohni S, Nemoto N (2012) Expression of somatostatin receptor (SSTR) subtypes (SSTR-1, 2A, 3, 4 and 5) in neuroendocrine tumors using real-time RT-PCR method and immunohistochemistry. Acta Histochem Cytochem 45:167–176
Ureshino N, Sueoka-Aragane N, Nakamura T, Sato A, Komiya K, Iwanaga K, Mitsuoka M, Takeda Y, Hayashi S, Sueoka E, Kimura S (2011) A fully integrated, automated and rapid detection system for KRAS mutations. Oncol Rep 26:609–613
Ureshino N, Aragane N, Nakamura T, Ide M, Mochinaga S, Fukushima N, Hayashi S, Sueoka E, Kimura S (2011) A fully integrated and automated detection system for single nucleotide polymorphisms of UGT1A1 and CYP2C19. Oncol Res 19:111–114
Nakamura T, Sueoka-Aragane N, Iwanaga K, Sato A, Komiya K, Abe T, Ureshino N, Hayashi S, Hosomi T, Hirai M, Sueoka E, Kimura S (2011) A noninvasive system for monitoring resistance to epidermal growth factor receptor tyrosine kinase inhibitors with plasma DNA. J Thoracic Oncol 6:1639–1648
Takeuchi K, Choi YL, Togashi Y, Soda M, Hatano S et al (2009) KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res 15:3143–3149
Takeuchi K, Choi YL, Soda M, Inamura K, Togashi Y, Hatano S, Enomoto M, Takada S, Yamashita Y, Satoh Y, Okumura S, Nakagawa K, Ishikawa Y, Mano H (2008) Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clin Cancer Res 15(14):6618–6624
Oken MM, Creech RH, Tormey DC, Horton J, Davis TE, McFadden ET, Carbone PP (1982) Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 5:649–655
Avruch J (2007) MAP kinase pathways: the first twenty years. Biochem Biophys Acta 1773:1150–1160
Ochi N, Takigawa N, Harada D, Yasugi M, Ichihara E, Hotta K, Tabata M, Tanimoto M, Kiura K (2014) Src mediates ERK reactivation in gefitinib resistance in non-small cell lung cancer. Exp Cell Res 322:168–177
Kubota Y, O’Grady P, Saito H, Takekawa M (2011) Oncogenic Ras abrogates MEK SUMOylation that suppresses the ERK pathway and cell transformation. Nat Cell Biol 13:282–291
Tetsu O, McCormick F (2003) Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell 3:233–245
Cragg MS, Kuroda J, Puthalakath H, Huang DC, Strasser A (2007) Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PloS Med 4:1681–1689
Hainsworth JD, Cebotaru CL, Kanarev V, Ciuleanu TE, Damyanov D, Stella P, Ganchev H, Pover G, Morris C, Tzekova V (2010) A phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with non-small cell lung cancer who have failed one or two prior chemotherapeutic regimens. J Thorac Oncol 5:1630–1636
Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, Parry J, Smith D, Brandhuber BJ, Gross S, Marlow A, Hurley B, Lyssikatos J, Lee PA, Winkler JD, Koch K, Wallace E (2007) Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res 13:1576–1583
Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios CH, Franke FA, Grinsted L, Smith PD, Zazulina V, Smith IC and Crino L (2012) Phase II double-blind, randomized study of selumetinib (SEL) plus docetaxel (DOC) versus DOC plus placebo as second-line treatment for advanced KRAS mutant non-small cell lung cancer (NSCLC). J Clin Oncol 30: suppl; abstr 7503
Sholl LM (2015) Biomarkers in lung adenocarcinoma: a decade of progress. Arch Pathol Lab Med. 139:469–480
Devarakonda S, Morgensztern D, Govindan R (2015) Genomic alterations in lung adenocarcinoma. Lancet Oncol 16(7):342–351
Klempner SJ, Ignatius Ou SH, Costa DB, VanderLaan PA, Sanford EM, Schrock A, Gay L, Ali SM, Miller VA (2015) The clinical use of genomic profiling to distinguish intrapulmonary metastases from synchronous primaries in non-small-cell lung cancer: a mini-review. Clin Lung Cancer 16(5):334–339
Acknowledgments
This work was supported by Nihon University School of Medicine 50th Anniversary Fund Research Grant (2012–2013).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tsujino, I., Nakanishi, Y., Hiranuma, H. et al. Increased phosphorylation of ERK1/2 is associated with worse chemotherapeutic outcome and a poor prognosis in advanced lung adenocarcinoma. Med Mol Morphol 49, 98–109 (2016). https://doi.org/10.1007/s00795-015-0130-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00795-015-0130-3