
Abstract
To ascertain the saying “Everything is everywhere, but the environment selects”, it was imperative to find out the main factor influencing bacterioplankton composition at genus level of Kongsfjorden where was influenced both by glacier melting water and Atlantic water. Thus, bacterioplankton diversity was investigated using pyrosequencing. In addition, nutrients, chlorophyll a, in situ temperature and salinity were measured. There were seventeen of 33 identified genera with relative abundance > 0.1%. Redundancy analysis showed that 73.02% of bacterioplankton community variance could be explained by environmental parameters. Furthermore, most of the abundant genera demonstrated significant correlation with environment parameters revealed by correlation analysis. Moreover, phosphate, nitrate and Chl a concentration, and the abundance of top nine identified genera varied with water mass significantly as shown by analysis of variance. Our results supported the notion that environmental factors, especially water mass had significant effect on bacterioplankton distribution at genus level. Considering the high sensitivity to environmental change and low error rate in identification, bacterioplankton at genus level could be potential bio-markers for monitoring environmental changes.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Svalbard is located closely to the warming core area and experiencing sudden rise in temperature. In western Svalbard Area, ice-free period increased by 3.3 days per year, and the surface temperature increased 0.5°C during 1980–2010 (Kortscha et al. 2012). Kongsfjorden is considered as a local indicator for climate change, and it is an ideal natural laboratory in Arctic to study the impact of climate change on marine ecosystem. This fjord, located at Svalbard (79°N, 12°E), is influenced by both Atlantic Ocean and glacier waters. The outer part of Kongsfjorden is connected to the inflow of transformed Atlantic water, and variable climate signals in the West Spitsbergen Current have influenced this fjord (Hop et al. 2012; Walczowski et al. 2012; Wassmann et al. 2015). Meanwhile, the inner part of Kongsfjorden is strongly influenced by the connected glaciers (e.g. glaciers Kongsbreen, Midtre Lovénbreen, and Austre Lovénbreen where the glacier runoff is about 25 ± 5 km3 a−1) (Hagen et al. 2003; Karner et al. 2013; Schellenberger et al. 2014). In addition, the sea ice melting water showed its impact on the water mass of this fjord (Gerland and Renner 2007), and regular winter ice cover has been rare since 2005/2006 (Cottier et al. 2007).
Bacterioplankton are important microbial communities in Polar Region, which experience extreme temperature and seasonal light variations. Though the abundance of bacterioplankton decreased from low latitude to high latitude, it has been estimated approximately at 108–109 cells/L in Kongsfjorden (Jankowska et al. 2005; Wang et al. 2009; Møller et al. 2011). Bacterioplankton, the key member of marine food web, are important in the microbial loop for their metabolic pathway in biogeochemical processes, such as carbon, nitrogen and phosphorus cycles (Falkowski et al. 1998). About 19% particulate organic carbon and 36% particulate nitrogen were accounted by bacteria in Kongsfjorden (Zhu et al. 2016). They are sensitive to environment change; however, the variance of bacterioplankton abundance and diversity could also exert a sustained influence on the micro-environment they thrived in, through microbial food web and metabolic pathway (Doney et al. 2012; Sunagawa et al. 2015).
The factors that influenced the bacterioplankton community structure have been reported before. These factors included seasonal changes, phytoplankton bloom, organic matters, the concentration of nutrients, and water depths (De Corte et al. 2011; Sunagawa et al. 2015; Luria et al. 2016; Sinha et al. 2016; Jain et al. 2019; Underwood et al. 2019). In Kongsfjorden, Bacteroidetes were found to be the most abundant bacterial group in spring season (Piquet et al. 2015), whereas Gamma-proteobacteria and Bacteroidetes were the dominant members in summer season (Zeng et al. 2013). The core taxa of particle-attached bacteria are formed by the members of Verrucomicrobia and Bacteroidetes, whose community composition is strongly influenced by carbohydrate content of particulate organic matter. However, the core taxa of free-living bacteria are the members of Alpha-proteobacteria and Gamma-proteobacteria, whose community composition is influenced by glacial meltwater influx and particulate organic carbon (Jain et al. 2019).
In fact, the factors affecting free-living bacteria composition are more complicated than particle-attached bacteria (Jain et al. 2019). The free-living bacterial community structure at the location near to Kongsbreen was also influenced by nitrite, phosphate and silicate, in addition to the glacial meltwater influx and particle organic carbon (Jain and Krishnan 2017; Jain et al. 2019). Besides, few studies focused on the influence of environment change on lower bacterioplankton taxonomy level (genus). This study was aimed to depict a clear picture of the bacterioplankton community diversity and structure from different taxonomy levels, and identify the main factor influencing their composition through the analysis of correlations with environment parameters at genus level in Kongsfjorden, Arctic.
Materials and methods
Study area and sampling
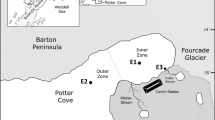
Samples were collected in Kongsfjorden, Ny-Ålesund (Svalbard, Norway, 78°56´N, 11°52´E) in July 2012 (Online Resource Fig. S1). A total of 14 samples from five stations were collected (Table 1). Sea water was collected using Niskin water bottle (KC-Denmark), and physical parameters (depth, temperature and salinity in situ) were determined with a Conductance–Temperature–Depth (SD204, SAIV A/S). For each sample, 2 L seawater was filtered using polycarbonate filters (47 mm diameter, 0.2 μm pore-size, Whatman), after prefilled by 3 μm pore size (47 mm diameter, Whatman). Samples were frozen at -80°C refrigerator immediately till further analysis.
Nutrients and Chlorophyll a determination
For each sample, 100 ml subsamples were filtered through 47 mm diameter Whatman GF/F filters, and the concentrations of inorganic nutrients (nitrate, nitrite, silicate and phosphate) were determined using autoanalyzer (Skalar San + + , Netherlands), following previous methods (Grasshoff et al. 1999; He et al. 2012).
In addition, 500 ml subsamples were filtered using 47 mm diameter Whatman GF/F filters under low vacuum pressure and the contents of chlorophyll a (Chl a) were determined using a fluorometer (Turner Designs 10, USA) following the method of Parsons (1984) and He et al. (2012).
Total DNA extraction, amplification and sequencing
Total DNA was extracted using the modified cetyltrimethylammonium bromide method (Cao et al. 2019), and analyzed by agarose gel electrophoresis. The bacteria 16S rDNA V1-V3 hyper-variable regions was amplified using universal primer pairs, F8 (5′-CCTATCCCCTGTGTGCCTTGGCAGTCTCAG-AGAGTTTGATCCTGGCTCAG-3′) (Turner et al. 1999) and R533 (5′-CCATCTCATCCCTGCGTGTCTCCGACTCAG-NNNNNNNN-TTACCGCGGCTGCTGGCAC-3′) (Sun et al. 2014) (the adaptor sequence were italicized and sample-specific barcode sequences were indicated as NNNNNNNN). Polymerase chain reaction (PCR) was performed using 5–10 ng of genomic DNA in a reaction volume of 20 μL, which contain 5 × FastPfu Buffer 4μL, 2.5mM dNTPs 2μL, forward primers (5 μM) 0.8 μL, reverse primer (5 μM) 0.8 μL, FastPfu Polymerase 0.4 μL, BSA 0.2μL and ddH2O up to 20 μL. The PCR conditions were as follows: initial denaturation at 95 °C for 2 min, 25 cycles as 95 °C for 30 s, 56.4 °C for 1 min, 72 °C for 30 s, and a final extension at 72 °C for 5 min. PCR products were purified using AxyPrep DNA purification Kit (Axygen, USA), and quantified with a Qubit® 2.0 Fluorometer (Life Technologies, USA). Pyrosequencing was performed using FLX Titanium Genome Sequencer (454/Roche Life Sciences, USA).
16S rDNA sequences data and statistical analyses
Mothur 3.1.2 (Schloss et al. 2009) was used to analyze 16S rDNA rough sequences. Reads were filtered as following parameters: length (400 ≤ length ≤ 650 bp), quality score (≥ 25), number of ambiguous bases (=0), and length of homopolymer runs (<8). Sequences were clustered at 97% similarity using Silva software (silva.seed_v119) (Pruesse et al. 2007) and used as reference sequences. Taxonomy was assigned to Operational Taxonomic Unit (OTU) according to Greengenes 13_5 database (McDonald et al. 2012). Sequences assigned as mitochondria and chloroplasts were deleted from the dataset for further analysis. Alpha-diversity indexes (ACE, Chao1, Shannon and invsimpson and Good’s coverage) were also determined using Mothur.
R (Version 3.1.2) was used to construct Venn diagram, and perform Detrended correspondence analysis (DCA), redundancy analysis (RDA) and partial redundancy analysis (pRDA) with the vegan package (Oksanen et al. 2018). Pairwise comparisons of environmental factors and bacterial communities were performed with a color gradient denoting of Pearson and Spearman’s correlation coefficients, including cluster analysis.
Analysis of variance (ANOVA) of bacterial communities and environmental factors among depth, stations and water masses, Pearson and Spearman analysis estimating significant differences and correlations between environment parameters and bacterial communities and within them, were performed using SPSS 20.0 software (IBM SPSS Statistics).
The sampling location map was prepared using ODV 4.5 (https://odv.awi.de.). The taxonomy composition pie chart and radar graph were constructed using Excel (Microsoft, USA). All figures in this paper were prepared using Illustrator CS4.0 (Adobe, USA).
Results
Water mass
Three water masses were identified based on in situ salinity and temperature data according to previous report (Svendsen et al. 2002). The high-temperature–low-salinity Surface Water (SW), characterized by temperature higher than 5°C and salinity ranging from 31.90 to 33.48 psu; the low-temperature–higher salinity Transformed Atlantic Water (TAW), characterized by temperature ranging from 2.47 to 3.64 °C and salinity higher than 34.68 psu; and the Transformed Intermediate Water (TIW), characterized by temperature ranging from 4.04 to 4.44 °C and salinity ranging from 34.39 to 34.57 psu (Fig. 1a; Table 1).

Distribution of temperature (°C), salinity (psu), water masses and nutrient in sampling locations. SW: T > 5°C, 31.90 > S > 33.48; TAW: 2.47 °C > T > 3.46 °C; S < 34.68 psu; TIW: 4.04 °C < T < 4.44 °C, 34.39 < S > 34.57. The dots represent the samples obtained, and the colors correspond to the depths on the color bar
The TAW, influenced by Atlantic water, was distributed from the deeper outer part toward the inner part of Kongsfjorden (deeper than 30 m); the TIW was mostly influenced by glacier runoff in summer, and distributed in the inner part of Kongsfjorden (K4-20 m and K5-20 m); and the SW was distributed throughout the surface of Kongsfjorden absorbing most of the sunlight.
Nutrients
Nutrient concentration varied with respect to sampling station, depth, and water masses (Fig. 1; Table 1). The average concentration of nitrate increased from SW (0.084 μmol L−1), TIW (0.135 μmol L−1) to TAW (0.199 μmol L−1). It also increased with the sampling depth with the highest concentration detected at 50 m of K1 station (K1-50 m, this abbreviation was used hereafter) (TAW) and lowest concentration detected at K1-0 m (SW) (Fig. 1b). Furthermore, the concentration of nitrite increased from inner to the outer part of Kongsfjorden with the minimum nitrite concentration detected at K4-0 m (SW) and maximum value was detected at K1-50 m (TAW) (Fig. 1c). Similarly, the phosphate concentration increased along the sampling depth with the maximum concentration detected at K4-50m (TAW), and minimum concentration at K1-0m (SW) (Fig. 1d). However, the concentration of silicate decreased from inner surface to the deeper and outer waters with the highest value detected at K4-0m (SW) and lowest value detected at K5-20 m (TIW) (Fig. 1e).
For Chl a, the average concentration was found to be 0.283 μg L−1 in summer Kongsfjorden. In contrast to nitrate and phosphate concentrations, Chl a concentration decreased from inner surface to the outer deeper waters of Kongsfjorden with the highest value detected at K4-0 m (SW) and lowest value at K2-50 m (TAW) (Fig. 1f).
Bacterioplankton diversity
In total, 34,644 sequences were classified into 1010 Operational Taxonomic Units (OTUs) after quality analysis and trimming of the original sequences at 97% similarity. The OTUs with single read were eliminated. Nearly half of the OTUs (46.06%) was found to be distributed throughout the three water masses. The unique OTUs in SW, TIW and TAW accounted for 9.70%, 2.48% and 17.13% of total OTUs, respectively (Fig. 2a). The mean reads of 14 samples were determined to be 2673 (from 1981 to 3389). The sequence data were submitted to the National Center for Biotechnology Information Sequence Read Archives under BioProject ID PRJNA299749.

Similarity in bacterioplankton composition among different water masses. a Venn diagrams representing the overlap of OTUs (at 3% sequence cutoff value) among water masses. b–d distribution of top bacterioplankton among water masses; the relative abundance was logarithmically transformed
The average good reads coverage was found to be 78.60% (75.53% to 80.04%). The highest alpha diversity index determined by ACE, Chao and Shannon were detected at K1-50 m, whereas the highest invsimpson index was detected at K5-0 m (Table 2). Minimum ACE and Chao1 index were detected at K3-0 m, and minimum Shannon and invsimpson indices were observed at K2-0 m.
Dominant bacterial communities
The most dominant bacterioplankton phylum was observed to be Proteobacteria, followed by Bacteroidetes, Actinobacteria, and Verrucomicrobia. Besides, Cyanobacteria, Firmicutes, Lentisphaerae, and groups OD1, SAR406, and TM7 were detected with minimal abundance (Online Resource Fig. S2).
Among the dominant bacterioplankton groups, Alpha-proteobacteria included the most abundance OTUs (45.7% of total OTUs), followed by Bacteroidetes (23.45%), Gamma-proteobacteria (10.88%) and Betaproteobacteria (7.82%). However, at the order level, Flavobacteriales (Bacteroidetes) were found to be the most abundant one (20.71% of total OTUs), followed by Rhodobacterales and Pelagibacterales (17.16% and 14.18% of the total OTUs, respectively) (Alpha-proteobacteria) (Online Resource Fig. S2a), each of the above order possessed more than 10% of total reads (34.78%, 19.20% and 18.44% of total reads, respectively) (Online Resource Fig. S2b). Another 12 orders containing more than 1% of total OTUs, were Caulobacterales (3.71%) of Alpha-proteobacteria, Burkholderiales (6.12%) of Betaproteobacteria, Oceanospirillales (4.59%), Alteromonadales (1.29%), Xanthomonadales (1.21%) of Gammaproteobacteria, Saprospirales (1.05%) of Bacteroidetes, Actinomycetales (1.45%) of Actinobacteria and Verrucomicrobiales (1.13%) of Verrucomicrobia. Moreover, the unclassified group of Alpha-proteobacteria (8.14%), Gamma-proteobacteria (2.90%), and other Bacteroidetes (1.69%) contained OTUs higher than 1% (Online Resource Fig. S2a).
Within total sequences, 53.38% was classified into 33 identified genera and 17 of them were detected at > 0.1% of the total sequences. At genus level, genera Pelagibacter and Octadecabacter of Alpha-proteobacteria were most detected with the relative abundance > 10% of the total sequences. The genera Ulvibacter (Bacteroidetes), Polaribacter (Bacteroidetes), Fluviicola (Bacteroidetes), Oceanibulbus (Alpha-proteobacteria) were detected with relative abundance between 1–10% of the total sequences, respectively. Similarly, Loktanella (Alpha-proteobacteria), Sediminicola (Bacteroidetes), Candidatus Portiera (Gamma-proteobacteria), MB11C04 (Verrucomicrobia), HTCC2207 (Gamma-proteobacteria), Candidatus Aquiluna (Actinobacteria), Pontirhabdus (Bacteroidetes), Erythrobacter (Alpha-proteobacteria), Pseudoruegeria (Alpha-proteobacteria), Xanthobacillum (Bacteroidetes), Coraliomargarita (Verrucomicrobia) were detected with the relative abundance of 0.1–1% of the total sequences.
The dominant taxa showed different distribution patterns in current study. In SW, some genera showed different trend from outer to inner of the Kongsfjorden. The abundance of Octadecabacter, MB11C04 and Pseudoruegeria increased from outer to inner of the Kongsfjorden (Fig. 2b, Table 3). Furthermore, the abundance of Pelagibacter and Candidatus Portiera was found to be decreased from outer to inner of the Kongsfjorden (Fig. 2b, Table 3), with the highest abundance of HTCC2207 detected at station K3 followed by station K2. In TAW, the genera Pelagibacter, Octadecabacter, Fluviicola and Sediminicola did not exhibit obvious difference among different sampling stations. For genera Ulvibacter and Polaribacter, the abundances in 30 m were higher than those of 50 m at K1 (Fig. 2c, Table 3). Ulvibacter biomass was found to be increased from outer to inner Kongsfjorden but Polaribacter biomass decreased at 30m and no obvious difference was detected at 50 m (Fig. 2c, Table 3). In TIW (K4-20m and K5-20m), five genera, Ulvibacter, Polaribacter, Fluviicola, MB11C04 and Xanthobacillum showed higher abundance in K5 station. Higher abundance of genera Pelagibacter, Octadecabacter, Candidatus Aquiluna, Erythrobacter, Pseudoruegeria and Coraliomargarita was observed at K4, while the other genera showed the similar abundance of K4 and K5 stations (Fig. 2d, Table 3). Furthermore, some genera with few reads were found to be unique to water mass. While Erythrobacter, Polaromonas and Thiobacillus were unique in SW, Fluviicola, Planktotalea and Rhodoferax were unique in TIW. Similarly, Luteolibacter and Leeuwenhoekiella et al. (15 genera) were unique in TAW (Online Resource Table S1).
Redundancy analysis (RDA) and partial redundancy analysis (pRDA)
The co-occurrence between environment and bacterioplankton communities was analyzed by RDA after DCA calculation (maximum axis length was 1.6026, which is < 3) (Lepx and Smilauer 2003).
RDA result indicated that 73.02% of the variance could be explained by environment parameters. Of these variances, 47.43% was caused by temperature, salinity and depth, 5.55% was caused by nutrients (nitrite, nitrate, ammonium, phosphate, silicate) and Chl a, and the rest (20.04%) could be attributed to the co-effect of temperature, salinity, depth, nutrients and Chl a (Table 4).
Analysis of variance (ANOVA)
The temperature, salinity, concentrations of nitrate, phosphate and Chl a were found to be changed significantly among the water masses (Table 1) as indicated by ANOVA results. Based on LSD method, multiple comparative analysis showed that both temperature and salinity of SW were significantly higher than those at other sampling depth. The nitrate concentration of SW and TIW were found to be significantly lower than that of TAW. Furthermore, phosphate concentration in SW was significantly lower than that in TAW, with the concentration in 20 m significantly lower than that in 50 m. Similarly, the concentration of Chl a in SW was significantly higher compared to that in TAW, with no significant differences observed among other depths (Table 1).
The abundance (total number of reads and OTUs of each sampling station) and alpha-diversity indices were not significantly varied among the sampling stations (Table 2). However, the number of OTUs and Chao index were significantly affected by sampling depth and water mass (Table 2) based on the ANOVA results. The number of OTUs and Chao index in 0 m were significantly lower than that in 20 m and 50 m. In addition, these two indices of SW showed significantly lower value than that of TAW and TIW. That may be caused by the nutrient difference.
For the dominant bacterioplankton genera, biomass varied with respect to sampling depth and water masses but not sampling stations (Table 3). There was significant difference among sampling depth and water masses for the distribution of genera, such as Ulvibacter and Fluviicola (>1%), Sediminicola, MB11C04, Pseudoruegeria and Coraliomargarita (each > 0.1%). However, only Ulvibacter exhibited significant difference among sampling stations (Table 3). Moreover, the distribution of genera Loktanella (>1%) showed significant difference among sampling depth, while Polaribacter (>1%) and Pontirhabdus (>0.1%) showed significant difference among water masses. Besides, none of the genera with abundance higher than 10% showed significant difference among sampling depth, stations and water masses.
Correlation analysis among environment factors and bacterioplankton communities
Spearman and Pearson analysis showed similar results as ANOVA. It was observed that temperature and salinity in situ (key points in determining the water masses), and sampling depth exhibited high impact on the distribution of nutrients and dominant bacterioplankton genera (Fig. 3; Online Resource Table S2 and S3). Sampling depth showed significant positive correlation with phosphate and nitrate, and negative correlation with Chl a, whereas sampling station exhibited significant negative correlation with nitrite (Online Resource Table S2 and S3). The genera, such as Ulvibacter, Polaribacter and Sediminicola, showed significant or very significant negative correlation with salinity and depth. However, they exhibited significant or very significant positive correlation with temperature. Only genera Octadecabacter (very significant) and Ulvibacter (significant) showed positive correlation with sampling stations (Online Resource Table S2 and S3). Besides depth and water masses, most nutrients exhibited significant correlation with bacterioplankton genera. Phosphate showed significant positive correlation with Oceanibulbus, significant negative correlation with Ulvibacter, and very significant negative correlation with Polaribacter and Sediminicola. Furthermore, only nitrite showed very significant negative correlation with Octadecabacter. Similarly, silicate exhibited no significant correlation with any bacterioplankton genera (Online Resource Table S2 and S3).

Pairwise comparisons of environmental factors and bacterioplankton genera abundance with a color gradient denoting Pearson (block) and Spearman’s (dot) correlation coefficients. The color and size indicate Pearson and Spearman correlation coefficients, respectively. Only significant correlations are shown (P < 0.05)
Discussion
Kongsfjorden, a well-known glacial fjord system, is influenced by the Atlantic Water. A higher temperature was observed in the current study (2.86–6.84 °C) compared to previously recorded data. The temperature in upper 50 m waters ranged from 2 °C to 4 °C in July 2000 (Svendsen et al. 2002), and from – 1 °C to 6 °C in July 2011 (Ji et al., 2014). There was an empirical model indicating that increasing 1 °C will induce a net balance of − 0.7 m and − 0.55 m w.e. for glaciers Austre Brøggerbreen and Midre Lovénbreen (Lefauconnier et al. 1993). Temperature is one of the major factors influencing prokaryotic life (Winter et al. 2008; Sunagawa et al. 2015), especially in low-temperature regions. Since the glacier runoff constituted about 5% of the fjord water mass (Cottier et al. 2005), high temperature indicates that glacier runoff will have a significant influence on Kongsfjorden bacterioplankton. Any small change in temperature might exhibit substantial changes in prokaryotic growth rates (Kirchman et al. 2009).
Chl a concentration was considered as an indicator for phytoplankton (Harding et al. 2016). The maximum average Chl a concentration in SW indicated relatively high biomass of phytoplankton, especially at K4-0 m and K5-0 m, which were mostly influenced by glacier runoff water. The growth of phytoplankton demonstrated that it consumed inorganic nutrients because relatively low nitrite, nitrate and phosphate values were observed in SW. Furthermore, the integration of terrestrial waters increased the silicate concentration, such as the water from Voitelva River (5.6 µmol L−1) and Wexelva Streamlet (11.8 µmol L−1) (Ji et al. 2014); however, phytoplankton in Kongsfjorden, such as diatom (the dominant group) would consume silicate in summer (Keck et al. 1999). This could explain the detection of higher concentration of Chl a and silicate; lower concentration of nitrite, nitrate and phosphate in SW. Moreover, the average concentration of nitrite, nitrate, phosphate and silicate were detected under the minimum threshold (inorganic nitrogen, 1.0 µmol L−1; phosphate, 0.1 µmol L−1; silicate, 2.0 µmol L−1), limiting the growth of phytoplankton (Justić et al. 1995).
The water masses can greatly influence the nutrients concentration. A year-round observation showed that a rapid and overwhelming intrusion of Atlantic Water across the shelf and into the fjord resulted into intense seasonal variance during midsummer (Cottier et al. 2005). The average Chl a concentration decreased from SW (0.468 µg L−1), TIW (0.250 µg L−1) to TAW (0.160 µg L−1) from inner surface water (K4-0 m and K5-0 m) to outer deeper water (K2-50 m which was influenced by Arctic Water). The average concentration of nitrate and phosphate increased from SW (0.084 µmol L−1; 0.728 µmol L−1), TIW (0.135 µmol L−1; 0.770 µmol L−1) to TAW (0.199 µmol L−1; 2.350 µmol L−1), respectively (Fig. 1b, d).
In this study, the bacterioplankton community composition was in agreement with the results obtained by Polymerase chain reaction-denaturing gel gradient electrophoresis (PCR-DGGE) and pyrosequencing, indicating that Alpha-proteobacteria, Bacteroidetes and Gamma-proteobacteria were the dominant clades in summer Kongsfjorden (Qiao et al. 2015; Zeng et al. 2013). Our results are consistent with the previous study investigated by DEEG (Zeng et al. 2009). These data of current study and previous researches suggested that there was no obvious variance of dominant bacterioplankton community composition annually at the phylum level. Although, it was reported that Gamma-proteobacteria was the most dominant clade followed by Bacteroidetes based on isolation method (Sinha et al. 2016). This difference would be caused by the selection of culture medium, in which the obtainable nutrition can stimulate or limit the growth of certain bacteria groups (Genevieve et al. 2019). With the development of culture-dependent methods, our understanding of bacterioplankton diversity and community composition comes to a new facet and a number of novel groups in marine environment have been revealed (Bowman et al. 2005; Zengler 2009). Meanwhile, the culture-dependent method provides more physiological and metabolic characteristics information about the bacteria isolations. Since there was no significant variance of dominant bacterioplankton composition at phylum level, the identified bacterioplankton genera with abundance of > 0.1% were considered using high-throughput sequencing and highly efficient annotation database. According to the “Everything is everywhere, but the environment selects” hypothesis, the abundance of identified genera exhibited different distribution trends among the identified water masses (Table 3). Octadecabacter, which had been reported from polar sea ice and water characterized by heterotrophic, psychrophilic, gas vacuolate (Gosink et al. 1997), showed relatively low abundance in TAW. That may be caused by the gas vesicles produced by the polar gas vacuolate strains, which help Octadecabacter rising up in the water column (Gosink et al. 1997). Meanwhile, the bacteria genera first reported from surface water or fresh water exhibited larger sequence numbers in SW, such as Oceanibulbus (>1%), Loktanella, and Sediminicola (Döbler et al. 2004; Trappen et al. 2004; Ivanova et al. 2005; Khan et al. 2006). Similar observation was made in a previous study in northern Baffin Bay (Fu et al. 2013). The structure of cultured Rhodobacterales was distinctive in different water masses where two ocean currents mixed in northern Baffin Bay (Fu et al. 2013).
The correlation analysis combined with environment parameters and the abundance of top 17 identified genera showed that 11 abundant identified genera exhibited significant correlation with at least two environment parameters. The abundance of Sediminicola, MB11C04, Pontirhabdus, Pseudoruegeria and Coraliomargarita showed significant correlation with phosphate and nitrate as demonstrated by Pearson and Spearman analysis. The genus Sediminicola can reduce nitrate (Khan et al. 2006; Hwang et al. 2015), so a significant negative correlation with nitrate was observed (Fig. 3). The genera Pontirhabdu and Coraliomargarita do not reduce nitrate to nitrite or nitrogen (Yoon et al. 2007; Yi et al. 2011; Zhou et al. 2019), and they show significant positive correlation with nitrate (Fig. 3). Though further research about how the nutrient influence the bacterioplankton abundance are in progress, our result has indicated that the nutrient concentration may promote/limit the abundance of the bacterioplankton genera. However, low-frequency significant correlation was observed among the abundant genera that only six appeared significant correlation with at least two abundant genera (Fig. 3, Online Resource Table S2 and S3). Results indicated that environment parameters highly influenced the bacterioplankton community structure than inter-correlation among bacterioplankton in summer Kongsfjorden. A previous study about Chukchi Borderland and surface water of Kongsfjorden also suggested that rare abundant phylotypes were apparently changed under environmental changes (Zeng et al. 2013). Though the abundance was only 0.1–1%, it can prove to be of great importance in the community functions (Hunter-Cevera et al. 2005).
Water mass also plays an important role in determining the bacterioplankton distribution and diversity at both order and genus level. The abundance and distribution of Rhodobacterales in an Arctic marine systems was determined by water mass (Fu et al. 2013). The abundance of 9 genera was significantly different among water masses according to ANOVA result using sequence data (Table 3). In TIW, the abundance of Octadecabacter and Fluviicola, which are typical sea-ice genera (Vollmers et al. 2013) and fresh water genus (O’Sullivan et al. 2005), was significantly higher than that in SW and TAW. However, the abundance of Pontirhabdus, first reported from sea water (Yi et al. 2011), was significantly higher in TAW than that in SW and TIW. This genus had not been observed in SW. Similarly, abundance of Ulvibacter and MB11C04 were significantly different among the three water masses.
Furthermore, the presence of unique OTUs in different water masses suggested their influence on bacterioplankton. The OTUs of genera Leeuwenhoekiella, Luteolibacter, and Coraliomargarita were detected only in TAW. Previous studies suggested that marine microbial communities in the Kongsfjorden–Krossfjorden system are shaped by both water mass origin (Atlantic, Arctic) and melting water input (Zeng et al., 2009; Piquet et al., 2011). The genera Leeuwenhoekiella (Nedashkovskaya et al. 2009), Luteolibacter (Jiang et al. 2012), and Coraliomargarita (Yoon et al. 2007; Zhou et al. 2019) are reported as isolated from surface water, fresh water or Arctic tundra. By transferring of melting water and being influenced by the Atlantic water, these genera came to deeper depth, such as TAW. When coupled with sampling depth, similar distribution was observed, exhibiting a visual influence of depth that was influenced by water masses. Our results supported the idea that water masses are determining the marine microbial communities at genus level. The increasing relative abundance of terrigenous bacteria, that an Alpha- and Gammaproteobacteria dominant community shifts to Cytophaga–Flavobacterium–Bacteroides-dominated community, further provided a secondary evidence that glacier runoff can influence bacterioplankton community structure (Piquet et al. 2011). The dominant bacteria phyla, Alpha-proteobacteria and Becteroidetes observed in this study are typical abundant bacteria in glacier or snow melting water (Larose et al. 2010; Zeng et al. 2013). Sphingomonadales and Actinomycetales are the abundant groups in current study, which have been reported from glacier and snow melting water with relative abundance of 16.4% and 7.7%, respectively (Larose et al. 2010).
Conclusions
Due to its susceptibility to environment change, Kongsfjorden is an ideal natural laboratory to study and monitor bacterioplankton community and diversity variance. On comparing the data from the current study and previous researches, there was no obvious difference of dominant bacterioplankton community composition annually at the phylum level. This study focused on the correlations of water masses and bacterioplankton communities at genus level. On combining the analysis of RDA, ANOVA, Pearson and Spearman, it can be suggested that environment factors, especially water mass exhibited significant influence on bacterioplankton abundance. Furthermore, the genera with relative abundance ranged from 0.1–1%, such as Pontirhabdus are more sensitive to water mass that could be developed as the candidate bio-markers for monitoring environmental changes. Further research is required to improve our understanding of organic carbon fluxes of the whole Svalbard glacier meltwater.
Abbreviations
- ANOVA:
-
Analysis of variance
- Chl a :
-
Chlorophyll a
- DCA:
-
Detrended correspondence analysis
- OTUs:
-
Operational taxonomic units
- PCR:
-
Polymerase chain reaction
- PCR-DGGE:
-
Polymerase chain reaction-denaturing gel gradient electrophoresis
- pRDA:
-
Partial redundancy analysis
- RDA:
-
Redundancy analysis
- SW:
-
Surface water
- TAW:
-
Transformed atlantic water
- TIW:
-
Transformed intermediate water
References
Bowman JP, McCammon SA, Dann AL (2005) Biogeographic and quantitative analyses of abundant uncultivated γ-proteobacterial clades from marine sediment. Microb Ecol 49:451–460
Cao SN, He JF, Zhang F, Lin L, Gao Y, Zhou QM (2019) Diversity and community structure of bacterioplankton in surface waters off the northern tip of the Antarctic Peninsula. Polar Res 38:3491 https://doi.org/10.33265/polar.v38.3491
Cottier FR, Nilsen F, Inall ME, Gerland S, Tverberg V, Svendsen H (2007) Wintertime warming of an Arctic shelf in response to large-scale atmospheric circulation. Geophys Res Lett 34:L10607
Cottier FR, Tverberg V, Inall ME, Svendsen H, Nilsen F, Griffiths C (2005) Water mass modification in an Arctic fjord through cross-shelf exchange: the seasonal hydrography of Kongsfjorden Svalbard. J Geophys Res 110:C12005
De Corte D, Sintes E, Yokokawa T, Herndl GJ (2011) Changes in viral and bacterial communities during the ice-melting season in the coastal Arctic (Kongsfjorden, Ny-Alesund). Environ Microbiol 13:1827–1841
Döbler IW, Rheims H, Felske A, El-Ghezal A, Flade-Schröder D, Laatsch H, Lang S, Pukall R, Tindall BJ (2004) Oceanibulbus indolifex gen. nov., sp. nov., a North Sea alphaproteobacterium that produces bioactive metabolites. IJSEM 54:1177–1184
Doney SC, Ruckelshaus M, Duffy JE, Barry JP, Chan F, English CA, Galindo HM, Grebmeier JM, Hollowed AB, Knowlton N, Polovina J, Rabalais NN, Sydeman WJ, Talley LD (2012) Climate change impacts on marine ecosystems. Annu Rev Mar Sci 4:11–37
Falkowski PG, Barber RT, Smetacek V (1998) Biogeochemical controls and feedbacks on ocean primary production. Science 281:200–206
Fu YY, Keats KF, Rivkin RB, Lang AS (2013) Water mass and depth determine the distribution and diversity of Rhodobacterales in an Arctic marine system. FEMS Microb Ecolo 84:564–576
Genevieve LF, Belle DS, Larissa DM, Ram MM, Samir RD (2019) Prokaryotic diversity in oxygen depleted waters of the Bay of Bengal inferred using culture-dependent and -independent methods. Indian J Microbiol 59:193–199
Gerland S, Renner AHH (2007) Sea-ice mass-balance monitoring in an Arctic fjord. Ann Glaciol 46:435–442
Gosink JJ, Herwig RP, Staley JT (1997) Octadecabacter arcticus gen. nov., sp. nov., and O. antarcticus, sp. nov., nonpigmented, psychrophilic gas vacuolate bacteria from polar sea ice and water. System Appl Microbiol 20:356–365
Grasshoff K, Kremling K, Ehrhardt M (1999) Methods of seawater analysis. Wiley-VCH, Weinheim, New York, Chichester, Brisbane, Singapore, Toronto
Hagen JO, Kohler J, Melvold K, Winther J (2003) Glaciers in Svalbard: mass balance, runoff and freshwater flux. Polar Res 22:145–159
Harding LW Jr, Mallonee ME, Perry ES, Miller WD, Adolf JE, Gallegos CL, Paerl HW (2016) Variable climatic conditions dominate recent phytoplankton dynamics in Chesapeake Bay. Sci Rep 6:23773
He JF, Zhang F, Lin L, Ma YX, Chen JF (2012) Bacterioplankton and picophytoplankton abundance, biomass, and distribution in the Western Canada Basin during summer 2008. Deep-sea Res Pt II 81–84:36–45
Hop H, Falk-Petersen S, Svendsen H, Kwasniewski S, Pavlov V, Pavlova O, Søreide JE (2012) Physical and biological characteristics of the pelagic system across Fram Strait to Kongsfjorden. Prog Oceanogr 71:182–231
Hunter-Cevera J, Karl D, Buckley M (2005) Marine microbial diversity: the key to earth’s habitability. American academy of microbiology, Washington DC
Hwang CY, Lee I, Cho Y, Lee YM, Jung YJ, Baek K, Nam S-II, Lee HK (2015) Sediminicola arcticus sp. nov., a psychrophilic bacterium isolated from deep-sea sediment, and emended description of the genus Sediminicola. IJSME 65:1567–1571
Ivanova EP, Zhukova NV, Lysenko AM, Gorshkova NM, Sergeev AF, Mikhailov VV, Bowman JP (2005) Loktanella agnita sp. nov. and Loktanella rosea sp. nov., from the north-west Pacific Ocean. IJSEM 55:2203–2207
Iversen RK, Seuthe L (2011) Seasonal microbial processes in a high-latitude fjord (Kongsfjorden, Svalbard): I. Heterotrophic bacteria, picoplankton and nanoflagellates. Polar Biol 34:731–749
Jain A, Krishnan KP (2017) Differences in free-living and particle-associated bacterial communities and their spatial variation in Kongsfjorden, Arctic. J Basic Microb. https://doi.org/10.1002/jobm.201700216
Jain A, Krishnan KP, Singh A, Thomas FA, Begum N, Tiwari M, Bhaskar VP, Gopinath A (2019) Biochemical composition of particles shape particle-attached bacterial community structure in a high Arctic fjord. Ecol Indic 102:581–592
Jankowska K, Wlodarska-Kowalczuk M, Wieczorek P (2005) Abundance and biomass of bacteria in two Arctic glacial fjords. Pol Polar Res 26:77–84
Ji ZQ, Gao SQ, Jin HY, He JF, Bai YC, Wang B, Yang Z, Chen JF (2014) Nutrient distribution and the influencing factors of seawater in Arctic Kongsforden, summer 2010. Acta Oceanologica Sincica 36:80–89
Jiang F, Li W, Xiao M, Dai J, Kan W, Chen L, Li W, Fang C, Peng F (2012) Luteolibacter luojiensis sp. nov., isolated from Arctic tundra soil, and emended description of the genus Luteolibacter. IJSEM 62:2259–2263
Justić D, Rabalais NN, Turner RE (1995) Stoichiometric nutrient balance and origin of coastal eutrophication. Mar Pollut Bull 30:41–46
Karner K, Obleitner F, Krismer T, Kohler J, Greuell W (2013) A decade of energy and mass balance investigations on the glacier Kongsvegen, Svalbard. J Geophys Res-Atmo 118:3986–4000
Keck A, Wiktor J, Hapter R, Nilsen R (1999) Phytoplankton assemblages related to physical gradients in an arctic, glacier-fed fjord in summer. ICES J Mar Sci 56:203–214
Khan ST, Nakagawa Y, Haryama S (2006) Sediminicola luteus gen. nov., sp. nov., a novel member of the family Flavobacteriaceae. IJSEM 56:841–845
Kirchman DL, Moran XA, Ducklow H (2009) Microbial growth in the polar oceans - role of temperature and potential impact of climate change. Nat Rev Microbiol 7:451–459
Kortsch S, Primicerio R, Beuchel F, Renaud PE, Rodrigues J, Lønne OJ, Gulliksen B (2012) Climate-driven regime shifts in Arctic marine benthos. PNAS 109:14052–14057. https://doi.org/10.1073/pnas.1207509109
Kwasniewski S, Gluchowska M, Walkusz W, Karnovsky NJ, Jakubas D, Wojczulanis-Jakubas K, Harding AMA, Goszczko I (2012) Interannual changes in zooplankton on the West Spitsbergen Shelf in relation to hydrography and their consequences for the diet of planktivorous seabirds. ICES J Mar Sci 69:890–901
Larose C, Berger S, Ferrari C, Navarro E, Dommergue A, Schneider D, Vogel TM (2010) Microbial sequences retrieved from environmental samples from seasonal arctic snow and meltwater from Svalbard, Norway. Extremophiles 14:205–212
Lefauconnier B, Vallon M, Dowdeswell J, Hagen JO, Pinglot JF, Pourchet M (1993) Global balance of Spitsbergen ice mass and prediction of its change due to climatic change. In I. Troen (ed): Symposium Global Change and Climate Change Impacts, Focusing on European Research. Copenhagen, 6–10 September 1993: EPOCH 0035 Scientific Report. European Programme on Climatology and Natural Hazards.
Lepx S, Smilauer P (2003) Multivariate analysis of ecological data using CANOCO. Cambridge University Press, Cambridge
Luria CM, Amaral-Zettleer LA, Ducklow HW, Rich JJ (2016) Seasonal succession of free-living bacterial communities in coastal waters of the western Antarctic Peninsula. Front Microbiol 7:1731. https://doi.org/10.3389/fmicb.2016.01731
McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P (2012) An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610–618
Moller AK, Barkay T, Abu Al-Soud W, Sorensen SJ, Skov H, Kroer N (2011) Diversity and characterization of mercury-resistant bacteria in snow, freshwater and sea-ice brine from the High Arctic. FEMS Microbiol Eco 75:390–401
Nedashkovskaya OI, Vancanneyt M, Kim SB, Zhukova NV, Han JH, Mikhailov VV (2009) Leeuwenhoekiella palythoae sp. nov., a new member of the family Flavobacteriaceae. IJSEM 59:3074–3077
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H (2018) Vegan: community ecology package, version. R package version 2.4.6. https://CRAN.R-project.org/package=vegan Accessed 20 May 2018
O'Sullivan LA, Rinna J, Humphreys G, Weightman AJ, Fry JC (2005) Fluviicola taffensis gen. nov., sp. nov., a novel freshwater bacterium of the family Cryomorphaceae in the phylum ‘Bacteroidetes’. IJSEM 55:2189–2194
Parsons TR (1984) A manual of chemical & biological methods for seawater analysis. Pergamon, Amsterdam
Pinhassi J, Bowman JP, Nedashkovskaya OI, Lekunberri I, Gomez-Consarnau L, Pedros-Alio C (2006) Leeuwenhoekiella blandensis sp. nov., a genome-sequenced marine member of the family Flavobacteriaceae. IJSEM 56:1489–1493
Piquet AM, Bolhuis H, Meredith MP, Buma AG (2011) Shifts in coastal Antarctic marine microbial communities during and after melt water-related surface stratification. FEMS Microbiol Eec 76:413–427
Piquet AMT, Maat DS, Confurius-Guns V, Sintes E, Herndl GJ, van de Poll WH, Wiencke C, Buma AGJ, Bolhuis H (2015) Springtime dynamics, productivity and activity of prokaryotes in two Arctic fjords. Polar Biol 39:1749–1763
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO (2007) SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35:7188–7196
Qiao ZZ, Zeng YX, Dong PY, Zheng TL (2015) Bacterial community composition and abundance of planktonic bacteria in Arctic Kongsfjorden in the summer of 2011. Chin J Polar Res 27:246
Schellenberger T, Dunse T, Kääb A, Kohler J, Reijmer C (2014) Surface speed and frontal ablation of Kronebreen and Kongsbreen, NW-Svalbard, from SAR offset tracking. Cryosphere Discuss 8:6193–6233. https://doi.org/10.5194/tcd-8-6193-2014
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microb 75:7537–7541
Sinha RK, Krishnan KP, Kerkar S, Divya DT (2016) Spatio-temporal monitoring and ecological significance of retrievable pelagic heterotrophic bacteria in Kongsfjorden, an Arctic Fjord. Indian J Microbiol 57:116–120
Sun FL, Wang YS, Wu ML, Jiang ZY, Sun CC, Chen H (2014) Genetic diversity of bacterial communities and gene transfer agents in northern South China Sea. PLoS ONE 9:e111892
Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, Djahanschiri B, Zeller G, Mende DR, Alberti A, Cornejo-Castillo FM, Costea PI, Cruaud C, d'Ovidio F, Engelen S, Ferrera I, Gasol JM, Guidi L, Hildebrand F, Kokoszka F, Lepoivre C, Lima-Mendez G, Poulain J, Poulos BT, Royo-Llonch M, Sarmento H, Vieira-Silva S, Dimier C, Picheral M, Searson S, Kandels-Lewis S, Tara Oceans coordinators, Bowler C, de Vargas C, Gorsky G, Grimsley N, Hingamp P, Iudicone D, Jaillon O, Not F, Ogata H, Pesant S, Speich S, Stemmann L, Sullivan MB, Weissenbach J, Wincker P, Karsenti E, Raes J, Acinas SG, Bork P (2015) Structure and function of the global ocean microbiome. Science 348:1261359
Svendsen H, Beszcynska-Moeller A, Hagen JO, Lefauconnier B, Tverberg V, Gerland S, Ørbæk JB, Bischof K, Carlo Papucci C, Zajaczkowski M, Azzolini R, Bruland O, Wiencke C, Winther JG, Dallmann W (2002) The physical environment of Kongsfjorden-Krossfjorden, an Arctic fjord system in Svalbard. Polar Res 21:133–166
Trappen SV, Mergaert J, Swings J (2004) Loktanella salsilacus gen. nov., sp. nov., Loktanella fryxellensis sp. nov. and Loktanella vestfoldensis sp. nov., new members of the Rhodobacter group, isolated from microbial mats in Antarctic lakes. IJSEM 54:1263–1269
Turner S, Pryer KM, Miao VP, Palmer JD (1999) Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J Eukaryot Microbiol 47:327–338
Underwood GJC, Michel C, Meisterhans G, Belzile C, Witt M, Dumbrell AJ, Koch BP (2019) Organic matter from Arctic sea-ice loss alters bacterial community structure and function. Nat Clim Change. https://doi.org/10.1038/s41558-018-0391-7
Vollmers J, Voget S, Dietrich S, Gollnow K, Smits M, Meyer K, Brinkhoff T, Simon M, Daniel R (2013) Poles Apart: Arctic and Antarctic Octadecabacter strains share high genome plasticity and a new type of Xanthorhodopsin. PLoS ONE. https://doi.org/10.1371/journal.pone.0063422
Walczowski W, Piechura J, Goszczko I, Wieczorek P, Notes A (2012) Changes in Atlantic water properties: an important factor in the European Arctic marine climate. ICES J Mar Sci 69(5):864–869. https://doi.org/10.1093/icesjms/fss068
Wang GZ, Guo CY, Luo W, Cai MH, He JF (2009) The distribution of picoplankton and nanoplankton in Kongsfjorden, Svalbard during late summer 2006. Polar Bio 32:1233–1238
Wassmann P, Kosobokova KN, Slagstad D, Drinkwater KF, Hopcroft RR, Moore SE, Ellingsen I, Nelson RJ, Carmack E, Popova E, Berge J (2015) The contiguous domains of Arctic Ocean advection: trails of life and death. Prog Oceanogr 139:42–65
Winter C, Moeseneder MM, Herndl GJ, Weinbauer MG (2008) Relationship of geographic distance, depth, temperature, and viruses with prokaryotic communities in the eastern tropical Atlantic Ocean. Microbl Eco 56:383–389
Yi H, Cho JC, Chun J (2011) Pontirhabdus pectinivorans gen. nov., sp. nov., isolated from seawater. IJSEM 61:2475–2481
Yoon J, Yasumoto-Hirose M, Katsuta A, Sekiguchi H, Matsuda S, Kasai H, Yokota A (2007) Coraliomargarita akajimensis gen. nov., sp. nov., a novel member of the phylum ‘Verrucomicrobia’ isolated from seawater in Japan. IJSEM 57:959–963. Zeng Y, Zheng T, Li H (2009) Community composition of the marine bacterioplankton in Kongsfjorden (Spitsbergen) as revealed by 16S rRNA gene analysis. Polar Bio 32:1447–1460
Zeng YX, Zhang F, He JF, Lee SH, Qiao ZY, Yu Y, Li HR (2013) Bacterioplankton community structure in the Arctic waters as revealed by pyrosequencing of 16S rRNA genes. Anton Leeuw Int J G 103:1309–1319
Zengler K (2009) Central role of the cell in microbial ecology. Microbiol Mol Biol Rev 73:712–729
Zhou LY, Wang NN, Mu DS, Liu Y, Du ZJ (2019) Coraliomargarita sinensis sp. nov., isolated from a marine solar saltern. IJSEM 69:701–707
Zhu ZY, Wu Y, Liu SM, Wenger F, Hu J, Zhang J, Zhang RF (2016) Organic carbon flux and particulate organic matter composition in Arctic valley glaciers: examples from the Bayelva River and adjacent Kongsfjorden. Biogeosciences 13:975–987. https://doi.org/10.5194/bg-13-975-2016
Acknowledgements
We give our thanks to all the members of Chinese Arctic Expedition 2012. This work was supported by the Chinese Polar Environment Comprehensive Investigation & Assessment Programs [CHNIARE-2011–2015], the National Natural Science Foundation of China [41206189 and 41476168], and Shanghai Natural Science Foundation [16ZR1439800]. Samples information and data were issued by the Resource-sharing Platform of Polar Samples (https://birds.chinare.org.cn), which was established by one of the National Science & Technology Infrastructures Polar Research Institute of China (PRIC) and Chinese National Arctic & Antarctic Data Centre (CN-NADC).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by H. Atomi.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cao, S., Zhang, F., He, J. et al. Water masses influence bacterioplankton community structure in summer Kongsfjorden. Extremophiles 24, 107–120 (2020). https://doi.org/10.1007/s00792-019-01139-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-019-01139-y