
Abstract
The present study investigates the purification and biochemical characterization of a novel extracellular serine alkaline protease, subtilisin (called SAPN) from Melghiribacillus thermohalophilus Nari2AT. The highest yield of protease (395 IU/g) with white shrimp shell by-product (40 g/L) as a unique source of nutriments in the growth medium was achieved after 52 h at 55 °C. The monomeric enzyme of about 30 kDa was purified to homogeneity by ammonium sulfate fractionation, heat treatment, followed by sequential column chromatographies. The optimum pH and temperature values for subtilisin activity were pH 10 and 75 °C, respectively, and half lives of 9 and 5 h at 80 and 90 °C, respectively. The sequence of the 25 NH2-terminal residues pertaining of SAPN exhibited a high homology with those of Bacillus subtilisins. The inhibition by DFP and PMSF indicates that this enzyme belongs to the serine proteases family. SAPN was found to be effective in the deproteinization (DDP %) of blue swimming crab (Portunus segnis) and white shrimp (Metapenaeus monoceros) by-products, with a degree of 65 and 82%, respectively. The commercial and the two chitins obtained in this work showed a similar peak pattern in Fourier-Transform Infrared (FTIR) analysis, suggesting that SAPN is suitable for the bio-production of chitin from shell by-products.
-
The purification of subtilisin (SAPN) from M. thermohalophilus Nari2A was carried out.
-
The molecular weight and the NH2-terminal sequence of the subtilisin were determined.
-
Optimum pH and temperature values for activity were pH 10 and 75 °C respectively.
-
SAPN was found to be effective in the deproteinization of crab and shrimp by-products.
-
SAPN may be used as candidate for chitin extraction from crustacean by-products.

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Proteases are paramount hydrolytic enzymes, widely used in the industrial sector, accounting for ~ 65% of the aggregate worldwide enzyme market, and ranking first enzymes before amylases and lipases. The market revenue of global protease sale is expected to reach $ 2.21 billion by 2021 (www.FreedoniaGroup.com) (Singh et al. 2016). Subtilisin proteases are a member of the subtilase superfamily, generally found in bacteria, archaea, and eukaryotic microorganisms. They are the most important industrial enzymes and essential bioadditives in modern laundry detergents in terms of boosting washing performance by removing efficiently insoluble proteinaceous stains (Vojcic et al. 2015). They are also used for the degradation of proteinaceous recalcitrant solid fish bio-waste, abundantly available in nature, by converting them into low value-added products, such as chitinous materials, protein hydrolysate, or amino-acids. In fact, fishery-processing industries generate large amounts of bio-wastes and non-edible parts (Lopes et al. 2018; Mao et al. 2017). The disposal of these by-products has always been considered as a serious issue, causing a major environmental and health problem. Of particular interest, cuticles of various crustaceans, especially crabs and shrimps, are the main sources of raw material for the production of chitin. This polymer, a long chain of N-acetyl-d-glucosamine residues linked by β-(1,4) bonds, is the most abundant renewable natural resource next to cellulose (Hammami et al. 2017). Owing to its numerous physico-chemical properties and biological activities, chitin is considered as an interesting bio-compound for several applications, with a great economic importance, including industrial, nutraceutical and medical sectors (Prameela et al. 2017; Wang et al. 2018). To date, a few studies on the use of proteolytic enzymes for the bio-deproteinization of shrimp wastes have been reported; however, little has been done with crab shell bio-wastes.
Blue swimming crab, P. segnis, and white shrimp, M. monoceros, belong to the order of Decapoda and are classified in the Portunidae and Penaeidae family, respectively. They are considered as two alien species in the Mediterranean Sea. P. segnis species is widespread in the west of the Atlantic Ocean. It proliferates easily and it has been introduced, recently, in the eastern Atlantic, in the north and east of the Mediterranean and Japan. Nowadays, it is found, extensively, in the Mediterranean, especially, in Tunisian coasts, causing various damages regarding fishing. M. monoceros species is native to the Red Sea; it has entered the Mediterranean via the Suez Canal and is now quite common in landings from the Gulf of Gabes in Tunisian coasts (Safaie et al. 2016).
The assessment of bacterial diversity and bioprospection of Chott Melghir salt lake located in the south-east of Algeria revealed the presence of two novel species of the genus Melghirimyces, for which the names of Melghirimyces algeriensis NariEXT, and Me. thermohalophilus Nari11AT were proposed and a novel species of a new genus, namely Me. thermohalophilus Nari2AT (Addou et al. 2013; Addou et al. 2012; Addou et al. 2015; Mohamed et al. 2019).
In this study, we present an overview on a recent research work in the field of subtilisin protease purification and characterization from the recently described polyextremophilic bacterium, Me. thermohalophilus Nari2AT, which also represented a novel genus. The potential relevance of deproteinization of crab and shrimp by-products to extract chitin is also discussed.
Materials and methods
Substrates, reagents, and proteases used in this work
Commercial chitin was purchased from P-Biomedical, France. Hammerstein casein was supplied by Merck (Darmstadt, Germany). The UNO Q-6 column (12 mm × 53 mm) was purchased from Bio-Rad Laboratories, Inc. (Hercules, CA, USA). The commercial proteases, subtilisin 309, trypsin, pepsin, and papain, were purchased from Sigma Chemical Company (St. Louis, MO, USA). Subtilisin 309 is active at pH 10, with a maximum activity at 60 °C. Trypsin functions at pH 8 with maximum activity at 40 °C. Pepsin is active at pH 2 and 50 °C. The optimum pH of papain is 7, while the optimum temperature was determined to be 65 °C. Alcalase™ Ultra 2.5 L and flavourzyme® 500 L were purchased from Novozymes Biopharma DK A/S (Bagsvaerd, Denmark). Alcalase™ Ultra 2.5 L is a commercial bacterial endo-protease of the serine type (subtilisin Carlsberg or subtilisin A). It is active between pH 6.5 and 8.5. It functions between 45 and 65 °C, with a maximum activity at about 60 °C. Flavourzyme® 500 L, produced by Aspergillus oryzae, is active at pH 7, with a maximum activity at 50 °C.
Preparation and protein analysis of P. segnis and M. monoceros shells powders
The blue swimming crab, P. segnis and white shrimp, M. monoceros shells by-products were obtained in fresh conditions from a fishery market located at Sfax, Tunisia. To obtain a fine powder, they were cooked at 100 °C for 20 min [1:2 ratio (w/v)] in distilled water, then desiccated and milled (Hamdi et al. 2017). After drying, they were stored at room temperature until further use. Total nitrogen content was measured by Kjeldahl method and the corrected protein concentration was obtained by subtracting chitin nitrogen from total nitrogen and multiplying by the factor 6.25 (Hamdi et al. 2017; Hammami et al. 2017).
Microorganism
Melghiribacillus thermohalophilus Nari2AT was previously isolated from the soil of Chott Melghir, an Algerian salt lake located at north-east of Biskra city, Algeria (34°00′00″–34°30′ 01″N 6°07′30″–6° 30′02″E), in the Saharan region. This strain is a novel thermophilic and a moderately halophilic bacterium. The temperature range for growth was (35–62 °C), with optimal growth occurring at 50–55 °C. The NaCl concentration range for growth was (0.5–17%, w/v), with an optimum at 7–10% (Addou et al. 2015). An attractive halo of casein degradation was revealed around the colony growth onto skimmed milk agar plates, after an incubation at 55 °C, as described previously (Mechri et al. 2017a, b). Strain Nari2AT was maintained at 4 °C on ISP 2 agar plates (Addou et al. 2015). The protease-producing isolate was propagated on ISP 2 agar plates at 55 °C and conserved in ISP 2 medium added to 20% glycerol stored at − 80 °C.
SAPN production
The pre-culture of Me. thermohalophilus Nari2AT was carried out in a 250 mL Erlenmeyer flask with baffles containing 25 mL of ISP 2 liquid medium composed of (g/L): glucose, 4; yeast extract, 4; malt extract, 4; CaCO3, 2; and 10% (w/v) NaCl then incubated at 55 °C overnight. This pre-culture was used to inoculate the culture, using the optimized medium at pH 7.4, containing only 40 g/L shrimp waste shells in Erlenmeyer flask with baffles.
The flasks were incubated at 55 °C for 52 h with shaking at 160 rpm in 500 mL Erlenmeyer flasks with a working volume of 50 mL. The obtention of clear supernatant crude protease solution and bacterial growth of strain Nari2AT were determined as described recently (Rekik et al. 2019).
Assay of proteolytic activity
As described previously by Mechri et al. (2017b) and Zaraî Jaouadi et al. (2012), the subtilisin activity was measured spectrophotometrically at 280 nm by the detection of aromatic amino-acids released from casein as a substrate in 100 mM glycine–NaOH supplemented with 2 mM CaCl2 (buffer A) at pH 10, after an incubation of 15 min at 75 °C.
SAPN purification
The SAPN production was undertaken using a fresh culture supernatant of strain Nari2AT, in the optimal medium. To remove microbial cells, 500 mL of a 52 h culture were centrifuged for 30 min at 10,000g. The supernatant was precipitated to 35% with solid (NH4)2SO4 and then re-centrifuged. Next, the clear supernatant was saturated to 60% (NH4)2SO4 and centrifuged; the pellet was dissolved in a minimal volume of 50 mM Tris–HCl buffer at pH 8 supplemented with 2 mM CaCl2 (buffer B). The clear supernatant was heated for 30 min at 80 °C. The insoluble denatured proteins were removed by centrifugation. The supernatant was loaded on a Sephacryl S-200 HR column (2.5 cm × 150 cm) pre-equilibrated with the buffer B. Protein contents (A280 nm) and subtilisin activity were determined. The pooled active fractions were then injected onto a UNO Q-6 column pre-equilibrated with 20 mM MOPS buffer at pH 7 supplemented with 2 mM CaCl2 (buffer C). The column was washed extensively with the same buffer. The proteins were eluted with buffer C containing a linear gradient of NaCl from 0 to 500 mM at a rate of 30 mL/h. Fractions of each peak were collected manually and estimated by measuring the absorbance at 280 nm and the protease activity on casein. Pooled fractions, containing SAPN, were concentrated for further analyses as detailed previously (Mechri et al. 2017a, b).
Analytical methods and proteins measurement
The total protein contents were determined using bovine serum albumin (BSA) as a standard. As described previously, three electrophoresis techniques, casein zymography, SDS-PAGE, and native-PAGE were used to analyze the SAPN purity, determine its molecular weight, and confirm its activity, respectively (Zaraî Jaouadi et al. 2012). The molecular weight was also confirmed by size exclusion HPLC, using a Shodex Protein WK 802-5 column (8 mm × 300 mm), pre-equilibrated with buffer A and native protein markers [in kDa] of 669: thyroglobulin (Rt = 9.150 min), 158: aldolase (Rt = 10.174 min), 43: ovalbumin (Rt = 11.067 min), 17: myoglobin (Rt = 13.800 min), and 1.35: vitamin B12 (Rt = 18.767 min).
Edman degradation
Bands of purified subtilisin SAPN separated on SDS gels were transferred to a ProBlott membrane (Applied Biosystems, Foster City, CA, USA), and the NH2-terminal sequence analysis was performed as detailed (Mechri et al. 2017b).
Physico-chemical characterization of the purified subtilisin SAPN
Influence of specific inhibitors and metallic ions on SAPN stability
Pre-incubation of SAPN with some specific inhibitors and reducing agents was investigated, as detailed recently (Rekik et al. 2019). The effects of different divalent metallic ions at an optimum concentration of 2 mM, on SAPN, were investigated by adding them to the reaction mixture after pre-incubating SAPN for 1 h at 40 °C.
Determination of the effects of pH on SAPN activity and stability
The full determination of pH on SAPN activity was investigated by measuring activity at 75 °C over a wide pH range from 3 to 13, using buffers at 100 mM each. The pH stability of SAPN enzyme was investigated by examining the residual activity after incubation of SAPN in different buffers with pH ranging from 8 to 11 at 40 °C for 24 h. The remaining subtilisin activity was then evaluated at the optimum pH as described above.
Determination of the effects of temperature on SAPN activity and stability
To study the effect of temperature on SAPN activity, an assay was conducted at temperatures ranging from 40 to 100 °C, at pH 10 with intervals of 5 °C. To evaluate its thermostability, SAPN was incubated for 24 h at temperatures, ranging from 60 to 90 °C, at pH 10, in the presence and absence of 2 mM CaCl2. Taking the non-heated SAPN as 100% activity, aliquots were withdrawn at desired time intervals to test the remaining activity.
Effect of osmolytes, KCl and NaCl, on SAPN protease activity
SAPN was pre-incubated for 1 h with various concentrations of KCl and NaCl from 0.5 to 10 M at pH 10. The activity of SAPN without osmolytes was taken as 100%. The residual subtilisin activity was measured under the standard conditions.
Determination of the effect of polyols on SAPN stability
Polyols are used for stabilization of enzymes in soluble form in enzymatic reactions; thus, various polyols, at 100 g/L, were tested to evaluate their effects on subtilisin stability at 80 °C in buffer A with a 1 h pre-incubation in each polyol. The activity of SAPN without any polyol and in the absence of calcium was taken as 100%. The residual protease activity was measured under the standard conditions.
Substrate specificity determination
As described previously, a number of natural, modified protein substrates as well as esters and synthetic peptides conjugated with p-nitroanilide (pNA) were used to investigate the substrate specificity profile of the subtilisin SAPN (Zaraî Jaouadi et al. 2013). The substrate, showing the highest activity was taken as the 100% reference.
Performance evaluation of the purified subtilisin SAPN
Kinetic measurements of SAPN, SPVP, SAPB, subtilisin 309, and subtilisin A proteases
The Michaelis constant (Km) and the maximum rate (Vmax) of the enzyme-catalyzed reaction were determined through a double reciprocal (Lineweaver Burk) plot of subtilisin activity towards casein, azo-casein, BTEE, and FAAF, as substrates at different concentrations, ranging from 0.10 to 50 mM at 50 °C and pH 10 for 10 min with Hyper32 program.
Determination of the degree of hydrolysis of proteins in crab and shrimp bio-wastes
The hydrolysis of proteins in crab and shrimp bio-wastes was carried out at 75 °C and pH 10 (for SAPN); 60 °C and pH 10 (for SPVP), 60 °C and pH 7 (for alcalase™ Ultra 2.5); 60 °C and pH 10 (for subtilisin 309); 50 °C and pH 7 (for flavourzyme® 500 L); 40 °C and pH 8 (for trypsin); 50 °C and pH 2 (for pepsin); and 65 °C and pH 7 (for papain). An amount of 2 g of each by-product was dissolved in 100 mL of assay buffer and then treated with 2000 U of commercial and purified enzymes. The pH was titrimetrically maintained constant using the pH–Stat set at the desired pH value of each enzyme by continuous addition of 4 N NaOH. The hydrolysis level was expressed as described previously (Hamiche et al. 2019).
Deproteinization of blue swimming crab blue and white shrimp bio-wastes by SAPN
The SAPN efficiency in the deproteinization of blue swimming crab (P. segnis) and shrimp (M. Monoceros) shells by-products was conducted using different enzyme/substrate [E/S] [unit of enzyme per milligram protein] ratios ranging from 0 to 30. The two shells homogenates (2 g) were mixed with 50 mL distilled water at 75 °C. The pH was adjusted to 10 with 5 N NaOH. The reactions were stopped by heating the solutions at 90 °C for 24 h to inactivate SAPN. The solid phase was washed and then pressed manually through four layers of gauze (Younes et al. 2014). The degree of deproteinization (DDP %) was calculated as described elsewhere (Hammami et al. 2017).
Chitin structural analysis with FTIR
Infrared spectra of commercial and extracted chitins were determined using a Nicolet FTIR spectrometer equipped with a horizontal attenuated total reflection (ATR) accessory. The internal crystal reflection was made from zinc selenide and had a 45° angle of incidence to the IR beam. The spectrum was acquired at a 4/cm resolution, and recorded between 4000 and 500/cm (mid-IR region). The data were analyzed by the OPUS 3.0 data collection software program (Bruker, Ettlingen, Germany).
Statistical analysis
All results derive from at least three independent replicates, and the control without proteolytic enzyme was under the identical standard experimental conditions. Results were expressed as the mean of the replicate determinations and standard deviation (± SD). Statistical significance and interpretation of results were estimated as detailed recently by the authors (Mechri et al. 2017a).
Results
Producing Nari2AT protease using powder crustaceans by-products
For the optimization of protease production by strain Nari2AT in the inexpensive medium with the powder of P. segnis or M. monoceros bio-wastes, a classical method “one-factor-at-a-time (OFAT)”, which involves changing one independent variable, while fixing others at certain levels, was adopted. Strain Nari2AT was cultured in Erlenmeyer flasks (500 mL) in an initial medium, containing 20 g/L of powder bio-waste from shrimp or crab for 52 h at 55 °C on a shaker incubator (160 rpm). Powder from shrimp by-product as the sole energy, carbon, and nitrogen sources gave an acceptable level of proteases production, with (7500 U/mL) followed by the powder from crab by-product with (3200 U/mL). Therefore, the effect of various concentrations of powder from white shrimp bio-waste on protease production by strain Nari2AT was studied. The optimum for the production of protease activity (15,800 U/mL) was obtained with 40 g/L of shrimp by-product with a significant yield of 395 IU/g of bio-waste (Fig. 1).

Time course of Me. thermohalophilus Nari2AT cell growth (empty triangle) and subtilisin production (filled triangle) using casein as substrate. Cell growth was monitored by measuring the absorbance at 600 nm and converted to cell dry weight (g/L). Each point represents the mean (n = 3) ± standard deviation
SAPN purification
The various purification steps are summarized in Table 1. The subtilisin activity recovery rate of the supernatant was 81%, even at a final ammonium sulfate concentration of 60%. By means of heat treatment, the specific activity of the SAPN enzyme increased by 6.4 times compared to that of crude enzyme. The clear supernatant obtained after heat treatment was loaded on a Sephacryl S-200 HR column. The fractions containing SAPN activity eluted at ~ 1.6 void volume (V0) were pooled together (data not shown). In the last step, the protein eluted at 160–200 mM NaCl from the UNO Q-6 FPLC column (Fig. 2a). Under optimum assay conditions, using casein as substrate, the purified SAPN had a specific activity of 94,800 U/mg, with a yield of 30%.

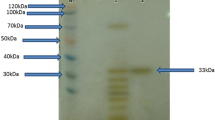
Purification of the protease SAPN from Me. thermohalophilus Nari2AT. a Chromatography profile of the purified subtilisin activity of SAPN enzyme on FPLC system using UNO Q-6. The column (15 mm × 68 mm) was equilibrated with buffer B. Adsorbed material was eluted with a linear NaCl gradient (0–500 mM) in buffer B at a flow rate of 60 mL/h, and assayed for protein content at 280 nm. b Assessment of homogeneity and molecular weight analysis of purified SAPN protein on native-PAGE, Lane 1, protein markers (molecular masses in kDa). Lane 2, purified SAPN enzyme from Me. thermohalophilus Nari2AT (30 μg). c Chromatography profile of SAPN on a Shodex Protein WK 802-5 column (8 mm × 300 mm), pre-equilibrated with buffer A. Proteins were separated by isocratic elution at a flow rate of 30 mL/h with buffer A. Protein peak, with a retention time (Rt) of 12.455 min, contained the protease activity. d 12% SDS-PAGE of the purified protease SAPN. Lane 1, Amersham LMW protein marker (GE Healthcare Europe GmbH, Freiburg, Germany). Lane 2, purified SAPN (30 µg) obtained after UNO Q-6 FPLC chromatography. e Zymogram caseinolytic activity staining. Purified subtilisin SAPN (30 µg)
Molecular weight determination of subtilisin SAPN
The purity of the purified SAPN enzyme eluted from the UNO Q-6 column (Fig. 2a) was checked by PAGE under non-reducing (Fig. 2b) and reducing (Fig. 2c) conditions and confirmed by gel filtration chromatography (Fig. 2d). The molecular mass of the SAPN enzyme was estimated to be about 30 kDa. This preparation was a homogeneous, as it exhibited a unique elution symmetrical peak at the retention time (Rt) = 12.455 min, corresponding to a single protein of nearly 30 kDa by HPLC gel filtration chromatography (Fig. 2c). The SDS-PAGE analysis of the pooled fractions showed one band, with an apparent molecular mass of about 30 kDa (Fig. 2d). The occurrence of only one clearance zone on the blue background in the zymography technique revealed the presence of caseinolytic activity and, therefore, of SAPN activity (Fig. 2e).
NH2-terminal amino-acid sequence determination of subtilisin SAPN
The unique sequence determined for the first 25 NH2-terminal amino-acids of the blotted purified SAPN confirmed that it was pure. This sequence was compared to protein sequences in the GenBank non-redundant protein database and the Swiss-Prot database, using the BLASTP and tBlastn search programs, respectively. As illustrated in Table 2, the NH2-terminal sequence of SAPN shared greatest sequence identity with Bacillus proteases, particularly those from B. licheniformis strain MP1 (88% identity with SAPN). Besides, it indicated 73 and 63% identities with proteases from Aeribacillus pallidus strain VP3 and Lysinibacillus fusiformis strain C250R, respectively (Table 2).
Physico-chemical characterization of purified subtilisin SAPN
Effects of inhibitors and metallic ions on stability of subtilisin SAPN
The profile of natural and synthetic inhibitors at an [inhibitor/enzyme] molar ratio = 100, as well as chelating agents and group-specific reagents, which affect the activity of subtilisin SAPN, are shown in Table 3. The SAPN activity was strongly inhibited by DFP and PMSF, eminent inhibitors of serine proteases, indicating that SAPN enzyme belongs to that subclass. In contrast, chymotrypsin and trypsin alkylating agents, as well as acid-, thiol-, zinc-, and cysteine-protease inhibitors had almost no effect on the activity of the purified SAPN. In this work, in the presence of 10 mM EDTA and 1 mM EGTA, SAPN retained, respectively, 67 and 86% of its activity, which suggests that no metal cofactors were required.
Table 3 also comprises the list of various metallic ions that were tested for an effect on SAPN activity. The addition of Ca2+ increased SAPN activity by 300%. Surprisingly, Cu2+ and Fe2+ increased the proteolytic activity, respectively, by 140 and 260%. SAPN activity was essentially unaffected by Mg2+ and Mn2+. Among all the tested metallic ions, only Hg2+, Co2+ and Cd2+ completely inhibited the purified protease SAPN.
Effects of pH on subtilisin activity and stability
The pH effect on the SAPN protease activity was determined in the range of pH 3–13, using casein as a substrate under the standard assay condition (Fig. 3a). The enzyme exhibited activity at all of the tested pH values, with a maximum activity at pH 10.

Physico-chemical proprieties of the purified SAPN from Me. thermohalophilus Nari2AT. Effects of pH (a) and the temperature (b) on the activity of SAPN. Effects of the pH stability (c) and thermostability (d) of SAPN. The subtilisin was pre-incubated in the presence and absence of CaCl2 at 60, 70, 80, and 90 °C. The activity of the non-heated subtilisin was taken as 100%. Each point represents the mean of three independent experiments. Each point represents the mean of three independent experiments. e Stability of SAPN in the presence of various polyols at 100 g/L. Subtilisin activity of the control sample, without additive, was incubated under similar conditions, and taken as 100%. Vertical bars indicate standard error of the mean (n = 3). f Effect of the thermostability of SAPN at 80 °C. The enzyme was pre-incubated in the absence (empty triangle) or presence of additive: 2 mM Ca2+ (filled triangle); 100 g/L PEG 1000 (filled diamond); and 2 mM Ca2+ and 100 g/L PEG 1000 (filled circle). The residual protease activity was determined from 0 to 24 h at 2 h intervals. The activity of the non-heated subtilisin was considered as 100%. Each point represents the mean (n = 3) ± standard deviation. Effect of NaCl and KCl (g) from 0.5 to 10 M on the stability of the purified SAPN
The effect of pH on the stability of SAPN was studied at 75 °C. Figure 3c indicates that the stability of the SAPN activity is very high in a wide pH range.
Effects of temperature on subtilisin activity and stability
To determine the temperature at which the purified enzyme SAPN showed optimum activity, the protease activity was monitored at various temperatures viz., 40, to 100 °C, as described in Fig. 3b. The enzyme showed activity at all the tested temperature values, and the maximum activity was obtained at 75 °C, in the presence of 2 mM Ca2+. Without 2 mM Ca2+, SAPN was optimally active at 65 °C.
For investigating the thermal stability of the purified protease, it was incubated at temperatures between 60 and 90 °C, as depicted in Fig. 3d. The enzyme did not display significant activity loss at the end of the 3 h incubation, with and without 2 mM Ca2+. A slight decrease in activity was determined after 4 h of incubation at 90 °C; the protease activity was reduced to 65 and 25%, respectively, with and without Ca2+.
Effect of polyols on the thermal stability
Among the polyols examined, the best results were obtained with PEG 1000 (Fig. 3e). Furthermore, SAPN thermostabilization was more effective with 2 mM calcium and PEG 1000 at 100 g/L, since the t1/2 at 80 °C was determined to be, respectively, 13 h, compared to 7 h in the absence of any additive (Fig. 3f).
Effect of KCl and NaCl on the thermal stability
The effect of osmolytes on the enzyme stability was studied increasing NaCl and KCl concentrations from 0 to 10 M (Fig. 3g). The optimal residual activity was reached at 5 M NaCl and 4 M KCl and was about 100%. Beyond 5 M NaCl, protease activity decreased progressively and retained 40% of its relative activity at 10 M NaCl. The relative activities of SAPN at 5 M KCl were about 22% of initial activity.
Substrate specificity
As given in Table 4, SAPN protease reacted with all proteins tested. It was able to hydrolyze casein, albumin, gelatin, ovalbumin, and keratin to varying degrees. However, it was not able to hydrolyse collagen type I and collagen type II. Furthermore, SAPN was not able to hydrolyze BAEE, TAME, and BCEE. Besides, it has a high hydrolysis rate towards BTEE and ATEE.
Synthetic peptide substrates, conjugated with p-nitroanilide, were also used to determine the pattern of specificity. The highest specificity of the protease was observed towards Suc-F-A-A-F-pNA followed by Suc-A-A-P-F-pNA. In addition, the relative activity of SAPN enzyme is 45% with Suc-A–A-P-L-pNA and 63% with Suc-L-L-V-Y-pNA when alanine and proline residues are present in P1 position. SAPN exhibited no detectable activity on Ac-Y-V-A-D-pNA.
Performance evaluation of the purified subtilisin
Determination of kinetic parameters
The kinetic parameters for casein, azo-casein, BTEE, and FAAF hydrolysis were determined under optimal experimental conditions for each enzyme, according to the Michaelis–Menten equation using Lineweaver–Burk plot.
The kcat/Km value of each enzyme (SAPN, SAPB, SPVP, subtilisin 309, and subtilisin A) is illustrated in Table 5. In the present report, SAPN showed a maximum turnover number (kcat) of 63,200/min for casein and a low Km of 0390 mM at 75 °C and pH 10 (Table 5). Therefore, catalytic efficiency (kcat/Km) was found to be 162,051/min/mM.
SAPN subtilisin has relatively higher Km for casein, azo-casein, BTEE, and FAAF. When casein was used as a substrate, SAPN was noted to exhibit kcat/Km values that were 2.6, 3.5, 9.7, and 17.1 times elevated than those of SPVP, SAPB, subtilisin 309, and subtilisin A, respectively. When azo-casein was used as a modified protein, SAPN was noted to exhibit kcat/Km values that were 3.3, 6.3, 10.9, and 13.7 times elevated than those of SPVP, SAPB, subtilisin 309 and subtilisin A, respectively.
A kinetic study was also performed using FAAF to investigate the effects of amino-acid residues adjacent to phenylalanine as a specific tetra-peptide substrate for subtilisin. FAAF was also the preferred substrate for SAPN with kcat/Km that were at least 2.0, 4.6, 12.8, and 18.4 fold higher, respectively, than those observed for SPVP, SAPB, subtilisin 309, and subtilisin A.
Determination of the degree of hydrolysis of the proteins in the by-products
The potential protein hydrolysis ability of each commercial and purified proteolytic enzyme, namely SAPN, SPVP, alcalase™ Ultra 2.5, subtilisin 309, flavourzyme® 500 L, trypsin, pepsin, and papain was tested by investigating the extent of hydrolysis. The hydrolysis curves of M. monoceros or P. segnis by-products, after 120 min of incubation, are shown in Fig. 4a, b. The purified SAPN from strain Nari2AT was the most efficient among the proteases. In fact, as shown in Fig. 4a, SAPN was the most efficient, with 34% protein hydrolysed used during hydrolysis of blue swimming crab P. segnis, followed by SPVP (24%), subtilisin 309 (17%), flavourzyme® 500 L (14%), alcalase Ultra 2.5 L (11%), papain (9%), trypsin (6%), and pepsin (4%). As shown in Fig. 4b, the purified SAPN was the most efficient, with 38% SAPN used during hydrolysis of white shrimp M. monoceros, followed by subtilisin 309 (19%), flavourzyme® 500 L (16%), alcalase Ultra 2.5 L (13%), papain (11%), and trypsin (8%), with pepsin being the least efficient (6%) one.

Performance evaluation of the purified subtilisin SAPN in M. monoceros and P. segnis proteins hydrolysis. a Hydrolysis curves of white shrimp M. monoceros treated with various purified and commercial proteases. The proteases used were: SAPN (filled diamond), SPVP (filled circle), subtilisin 309 (empty circle), flavourzyme® 500 L (filled square), alcalase™ Ultra 2.5 L (empty square), papain (filled triangle), trypsin (empty triangle), and pepsin (asterisk). Each point represents the mean (n = 3) ± standard deviation. b Hydrolysis curves of blue swimming crab, P. segnis proteins treated with the same purified and commercial proteases. Each point represents the mean (n = 3) ± standard deviation. c Effects of the E/S ratio on the deproteinization of white shrimp M. monoceros and blue swimming crab, P. segnis by-products. Protein hydrolysis was performed for 3 h at pH 10 and 75 °C. Each point represents the mean (n = 3) ± standard deviation
Chitin extraction from blue swimming crab blue and white shrimp bio-wastes
Deproteinization of swimming crab P. segnis or white shrimp M. monoceros by-products using SAPN from Nari2AT was first carried out at pH 10 and 75 °C. As shown in Fig. 4c, in the control experiments without SAPN, there was hardly any deproteinization of white shrimp and blue crab shells. The addition of 5 U of SAPN protease/mg proteins for white shrimp shells and blue swimming crab improved the efficiency of proteins removal and eliminated around 30 and 20% of proteins, respectively. However, the deproteinization ratio with E/S = 5 U/mg was 80%, and the percentage of protein removal increased with increasing E/S ratio, reaching about 93% with E/S ratio of 10 U/mg.
However, the percentage of protein removal increased with increasing E/S ratio and reached about 60 and 78% for blue swimming crabs and white shrimp shells, respectively, beyond a ratio of E/S = 20 U/mg. Additional increase in SAPN concentration did not improve significantly the deproteinization rate.
Chitin characterization by FTIR
The two extracted chitins were subjected to FTIR spectroscopy analysis, and compared with the commercial one, as shown in Fig. 5. The spectra of the P. segnis and M. monoceros samples exhibited typical α-chitin features, without bands at 1540/cm with bands above 3000/cm, with peaks at 2919, 2922, 1621, 1600, 1429, 1403, 1010, and 1040/cm.

FTIR analysis of chitins. a Commercial chitin. b Swimming crab chitin (CPs) treated with subtilisin SAPN. c White shrimp M. Monoceros chitin (CMm) treated with subtilisin SAPN
Discussion
Hypersaline environments constitute typical examples of environments with extreme conditions due to their exposure to high and low temperatures, high salinity, high pH values and in some cases, ultrasonic vibrations, and low oxygen conditions. In halophilic or halotolerant bacterial proteins, the ‘halo-adaptation’ is achieved by multiple ways. A novelty in their biochemical significance, structure and characteristics, and a thorough understanding of robust proteases, exceptionally salt-stable subtilisin proteases, denoted as halolysin functioning, could be of prime importance (Mokashe et al. 2018).
The application of fish processing by-products as cheap substrate for the production of proteolytic enzyme could also reduce environmental pollution. P. segnis crab powder and M. monoceros shrimp by-products are two essential sources for the development of the strain Nari2AT and the synthesis of its metabolites. Previously, a significant production was noted for the proteolytic enzymes by B. pumilus strain A1 and B. mojavensis strain A21 (1267 and 214 U/mL, respectively, on a medium containing 50 g/L powder of shrimp by-product) (Ghorbel Bellaaj et al. 2012). Similarly, Annamalai et al. documented a production of halo-stable proteases from halophilic bacterial strains using marine bio-wastes (Annamalai et al. 2014a, b).
The protease activity of SAPN is assayed and the protein concentration is determined. This relatively good recovery rate, indicated that ammonium sulfate precipitation is efficient in proteins precipitation (Amid et al. 2012). The high level of specific activity strongly suggested the potential prospects of SAPN in various industrial applications.
A variable molecular weight of bacterial serine proteases was determined; viz the alkaline protease from B. licheniformis MP1 with MW of 49 kDa (Jellouli et al. 2011) and the SPVP enzyme from Aeribacillus pallidus VP3 with MW of 29 kDa (Mechri et al. 2017a). On the other hand, a protease from Bacillus sp. ZJ1502 has an extremely low molecular weight of 14 kDa (Yu et al. 2019). Serine proteases that have larger molecular weight are also available in the literature. Taking the example of the one secreted from B. halotolerans strain CT2 had high molecular weight of 250 kDa (Dorra et al. 2018). These illustrated results potently suggest that SAPN protease from strain Nari2AT is a monomeric protein comparable to those previously reported for other proteases from Bacillus strains (Mechri et al. 2017b; Zaraî Jaouadi et al. 2012).
The NH2-terminal sequence of SAPN showed uniformity. It differed to B. licheniformis strain MP1 by 3 amino-acid residues at positions 14, 17, and 23. Moreover, it differed to subtilisin Novo from B. amyloliquefaciens strain DC-4 by ten amino-acid residues at positions T3S, I8V, P9S, L10Q, F14P, K15A, V16L, P17H, A18S, and F21Y. These results revealed that SAPN protease is a new subtilisin enzyme.
PMSF is known to sulphonate the essential serine residue on the active site of the SAPN enzyme, resulting in a decrease in enzyme inactivation (Rani et al. 2012). This group of salt-stable serine or subtilisin-like serine protease, denoted as ‘halolysin’, is isolated from different halophilic Bacillus and related groups such as Alkalibacillus sp. strain NM-Fa4, Oceanobacillus iheyensis strain O.M.A18, B. halodurans strain US193, and Halobacillus sp. strain SCSIO 20089 (Daoud et al. 2018; Mesbah and Wiegel 2014; Purohit and Singh 2014; Yang et al. 2013). The inhibition profile was similar to that observed for the purified SAPHM produced by the alkaline protease of B. licheniformis K7A (Hadjidj et al. 2018). Furthermore, aspartic and cysteine endo-proteases are rarely found in halophilic and halotolerant bacteria.
The moderate inhibition by EDTA and EGTA suggested that SAPN enzyme is not a metalloprotein. Similar result was reported for alkaline proteases recovered from Bacillus strains (Hadjidj et al. 2018; Mechri et al. 2017b). The stability of the enzyme in the presence of EDTA is advantageous for the use of SAPN as detergent additive. The fact that the proteolytic enzyme shows high stability in the presence of EDTA and EGTA is of great significance with respect to detergent industry use. In fact, the chelating agents such as EDTA and EGTA are among the most commonly found components in detergent formulations, used to soften water and assist in removing stains. The majority of enzymes of halophilic or halotolerant bacteria require the presence of Ca2+. In fact, two calcium-binding sites are present in other members of the subtilisin superfamily, including subtilisin SAPRH (Rekik et al. 2019). Metal-binding sites can function to both stabilize and activate enzymes. The inhibition of SAPN activity by cobalt and cadmium which are two heavy metals could be perhaps ascribed to their binding to catalytic residues in the active site of the enzymes. Such an observation was made for the purified protease KERUS strongly inhibited by cobalt, nickel, and mercury (Zaraî Jaouadi et al. 2013). Rekik et al. explored the engineering of binding affinity at metal ion binding sites for the stabilization of subtilisin conformation (Rekik et al. 2019). Rather, SAPB, purified from B. pumilus strain CBS, is strongly stabilized by calcium and magnesium ions at specific sites in its tertiary structure, preventing autolysis and thermal unfolding (Zaraî Jaouadi et al. 2012). It is worth noting, in this context that, considering potential industrial application, the activity of SAPN in the presence of metallic ions is a key property.
SAPN had a good activity in pH range from neutral to alkaline; however, the activity was higher in alkaline pH. In line with the current study, the serine protease from Lysinibacillus fusiformis C250R has an optimum pH at 10, as the same of SAPN (Mechri et al. 2017b). Proteases from Caldicoprobacter guelmensis D2C22T and B. licheniformis K7A also show an optimum of 10 (Bouacem et al. 2015; Hadjidj et al. 2018).
Bacterial proteases are active over a wide range of pH, depending on the source of the bacteria from which they have been isolated. The range of pH stability of SAPN from strain Nari2AT will be very useful in terms of industrial applications, particularly, in commercialized laundry detergents. An alkaline pH in the wash water can considerably improve the cleaning ability of the detergent. They are generally optimal in an alkaline pH range of 9–11. Other serine proteases with an optimum temperature of 75 °C have been reported in the literature. The optimum pH range of alkaline proteases is generally between 55 and 65 °C (Hadjidj et al. 2018), with a few exceptions at higher temperature optima of 70 and 80 °C (Bouacem et al. 2015; Mechri et al. 2017a; Omrane Benmrad et al. 2018). All the results obtained strongly suggested the high thermostable nature of the enzyme, thus ideal for the applications in alkaline environments. Results of the study that was performed demonstrated that the SAPN protease is much more stable at 70 and 80 °C than the serine protease SAPRH purified from B. safensis strain RH12 (Rekik et al. 2019).
The addition of each polyol increased the stability of SAPN from Nari2AT towards 80 °C after pre-incubation at 1 h over the control. Rather, literature studies have confirmed that the addition of substances, such as polyols, enhanced the stability of several proteolytic enzymes (Omrane Benmrad et al. 2018; Shakilanishi et al. 2017). In fact, studies have been conducted, revealing that the effectiveness of PEG on proteins highly depends on the polymer molecular weight and on the protein structure. Moreover, PEG with high molecular weights is believed to prevent protein–protein interactions, leading to stabilization (Nirmal and Laxman 2014). A thorough study of the process of deactivation pertaining to proteases has shown that improving thermal stability is essential for efficient operation in numerous industrial applications (Mechri et al. 2017b; Rekik et al. 2019). The increase in the thermal stability by adding polyols was probably due to the reinforcement of the hydrophobic interactions among nonpolar amino-acids inside the enzyme molecules, thus increasing their resistance to inactivation. The investigation of SAPN substrate specificity is interesting, because no quantitative measure of absolute substrate specificity exists. Rather, specificity must be discussed in relative terms, wherein ratios of catalytic parameters with multiple substrates are presented to ascertain patterns of reactivity (Li et al. 2018). Purified proteases with high specificity for substrates can be used in industrial processes in such similarly to chemical catalysts, and their recovery and reuse would facilitate a better cost–benefit ratio. The specificity of proteolytic enzyme modulated by the classification of amino-acids as well as by functional group, aromatic, aliphatic or sulphur-containing, determines the specificity of the enzyme to the substrate close to the bond being hydrolysed (Hadjidj et al. 2018). In fact, subtilisin that preferentially hydrolyzes peptide links, contributed by hydrophobic (Ala, Val, Leu, Ile, Met, Phe, Tyr, and Trp), cationic (Lys and Arg), anionic (Asp and Glu), and polar (Ser and Gln) side chains at P1 as well as proline at P1, which are known to exist. As expected, among natural and modified proteins, the casein and azo-casein were hydrolysed to a much higher degree than the albumin, gelatin, ovalbumin, keratin, albumin azure, and keratin azure, which is in accordance with other investigations of substrate specificity profiles of subtilisins (Rekik et al. 2019). In fact, they are found to be more active against casein compared to albumin, ovalbumin, and keratin, since albumin and ovalbumin are present in several stains. These results indicate the utility of the purified enzyme SAPN for detergent formulation. Thus, it can also be used in pharmaceutical and cosmetic purposes, or in the leather industry, where collagen should not be attacked (Zaraî Jaouadi et al. 2013). SAPN was noted to exhibit both esterase and amidase activities on N-terminal and C-terminal protected l-tyrosine. This enzyme has no esterase and amidase activities on N-terminal and C-terminal protected l-arginine and cysteine. This behavior has already been described for other subtilisins from Bacillus origins towards esters (Hadjidj et al. 2018; Mechri et al. 2017b). In contrast to these results, another serine keratinolytic protease, KERUS, did not show protease activity towards ATEE and BTEE. Several biologically active peptides are broken by the enzyme at sites consistent with the specificity deduced from studies with model synthetic substrates. The higher subtilisin activity with Suc-F-A-A-F-pNA and Suc-A-A-P-F-pNA substrates shows that the SAPN enzyme specifically prefers hydrophobic substrates, with aromatic residues occupying the P1 and P4 positions. Mechri et al. (2017a) reported that another serine protease, SPVP, showed similar activity towards Suc-F-A-A-F-pNA and Suc-A-A-P-F-pNA, while it did not show activity towards Suc-A-A-A-pNA and a very weak activity towards Suc-A-A-V-pNA, which suggested that SPVP protease largely preferred hydrophobic substrates, with aromatic residues occupying the P1 and P4 positions of pNA substrates. In contrast to these results, other serine keratinolytic proteases, such as KERUS and KERCA and KERQ7, did not show protease activity towards Suc-F-A-A-F-pNA and Suc-A-A-P-F-pNA (Zaraî Jaouadi et al. 2013). Proline at the P2 position was also noted to promote hydrolysis by SAPN enzyme, a feature that was not observed for other subtilisins. Hence, the substrate specificity profiles of the proteolytic enzyme SAPN were closely similar to subtilisins, not only in terms of specificity for position P1, but also with regard to the effects of amino-acid residues neighboring the cleavage site.
The shape of the hydrolysis curve (Fig. 4) is similar to those previously published for cuttlefish muscle, keratin, and casein (Balti et al. 2015; Hamiche et al. 2019; Omrane Benmrad et al. 2016). These findings indicate that SAPN can be useful for the preparation of protein and peptides hydrolysates. In fact, an optimal release of protein from blue swimming crab P. segnis or white shrimp M. monoceros while leaving chitin intact is a promising industrial and biotechnology opportunity. The quality of chitin depends on the extraction process. In the cuticles of crustaceans, chitin is closely associated with lipids, proteins, and minerals (Arbia et al. 2013). These compounds should be removed to achieve the purity degree of chitin, necessary for biological and industrial applications (Younes et al. 2016). Chitin from crustaceans can be recovered chemically or enzymatically. Since chemical deproteinization results in high amounts of bases and acids waste, enzymatic deproteinization using proteolytic enzymes is considered as the most favorable approach and an alternative treatment to alkali digestion (Doan et al. 2019; Dun et al. 2019).
Despite proteins, probably related to chitin by covalent bonds, which require a chemical or enzymatic treatment for their recovery, some proteins may be associated with the chitinous matrix by electrostatic forces or hydrogen bonds that can be dissociated by heat treatment (Hamdi et al. 2017; Jellouli et al. 2011). Other proteins are probably linked to chitin by hydrogen bonds and electrostatic forces, whose removal requires enzymatic treatment or additional chemical.
The ability of the subtilisin SAPN to rapidly and efficiently degrade recalcitrant solid by-product may be harnessed for developing an eco-friendly and cost-effective process for solid bio-waste management, especially from crustacean shells. The absence bands at 1540/cm proved the efficacy of enzymatic deproteinization, and thereby, the purity of the extracted chitin (Lassoued et al. 2015). In the stretching vibration region of OH and NH, groups above 3000/cm are assigned to the vibrational modes involved in intermolecular hydrogen bonding CO–HN and the intramolecular bonds –NH groups, respectively (He et al. 2012). The peaks at 2919 and 2922/cm were assigned to the stretching vibration of –CH3 and –CH2, respectively (Hamdi et al. 2017). In addition, the splitting of the amide I band in the blue swimming crab and shrimp chitins spectra was observed, with the occurrence of peaks at 1621 and 1600/cm, attributed to the intermolecular hydrogen bonds CO–HN and at 1555/cm due to the intramolecular hydrogen bond CO–HOCH2 (Liu et al. 2012; Mohammed et al. 2013). Rather, a band at 1429/cm, attributed to CH2 group occurred in the FTIR spectra of the extracted chitin from shrimp by-product. The peaks at 1403, 1010, and 1040/cm for the characteristic peaks of β-(1-4)-glucoside bond in chitin are both shown in Fig. 5a–c, which demonstrated that the enzymatic deproteinization of the two bio-wastes by SAPN did not destroy the structure of glucoside molecule (He et al. 2012).
Conclusions
This study describes in this study allowed the production, purification and characterization of an extracellular protease secreted by Me. thermohalophilus strain Nari2AT, aiming at its utilization in the production of chitin from blue swimming crab, P. segnis, and white shrimp, M. monoceros. The process developed in this investigation allowed to produce a novel subtilisin with interesting properties in a process with alternate substrates from crustacean’s by-products, thus, creating an outstanding alternative to managing them. Moreover, these investigations inspired us to explore the enzymatic biodiversity of other soil microorganisms of Chott Melghir. Further works are needed to be translated from laboratory and fundamental experiment to pilot an industrial-scale application. Therefore, the extracellular overproduction of SAPN, its thermostable nature, and its use in deproteinization of crab and shrimp shell wastes may find potential applications in the valorization of shrimp and crab bio-wastes to produce chitin.
References
Addou AN, Schumann P, Spröer C, Hacene H, Cayol JL, Fardeau ML (2012) Melghirimyces algeriensis gen. nov., sp. nov., a member of the family Thermoactinomycetaceae, isolated from a salt lake. Int J Syst Evol Microbiol 62:1491–1498
Addou AN, Schumann P, Spröer C, Bouanane Darenfed A, Amarouche Yala S, Hacene H, Cayol JL, Fardeau ML (2013) Melghirimyces thermohalophilus sp. nov., a thermoactinomycete isolated from an Algerian salt lake. Int J Syst Evol Microbiol 63:1717–1722
Addou NA, Schumann P, Spröer C, Hania WB, Hacene H, Fauque G, Cayol JL, Fardeau ML (2015) Melghiribacillus thermohalophilus gen. nov., sp. nov., a novel filamentous, endospore-forming, thermophilic and halophilic bacterium. Int J Syst Evol Microbiol 65:1172–1179
Amid M, Shuhaimi M, Islam Sarker MZ, Abdul Manap MY (2012) Purification of serine protease from mango (Mangifera Indica Cv. Chokanan) peel using an alcohol/salt aqueous two phase system. Food Chem 132:1382–1386
Annamalai N, Rajeswari MV, Balasubramanian T (2014a) Extraction, purification and application of thermostable and halostable alkaline protease from Bacillus alveayuensis CAS 5 using marine wastes. Food Bioprod Process 92:335–342
Annamalai N, Rajeswari MV, Sahu SK, Balasubramanian T (2014b) Purification and characterization of solvent stable, alkaline protease from Bacillus firmus CAS 7 by microbial conversion of marine wastes and molecular mechanism underlying solvent stability. Process Biochem 49:1012–1019
Arbia W, Arbia L, Adour L, Amrane A (2013) Chitin extraction from crustacean shells using biological methods: a review. Food Technol Biotech 51:12–25
Balti R, Bougatef A, Sila A, Guillochon D, Dhulster P, Nedjar Arroume N (2015) Nine novel angiotensin I-converting enzyme (ACE) inhibitory peptides from cuttlefish (Sepia officinalis) muscle protein hydrolysates and antihypertensive effect of the potent active peptide in spontaneously hypertensive rats. Food Chem 170:519–525
Benkiar A, Zaraî Jaouadi N, Badis A, Rebzani F, Boulkour Touioui S, Rekik H, Naili B, Ferradji FZ, Bejar S, Jaouadi B (2013) Biochemical and molecular characterization of a thermo- and detergent-stable alkaline serine keratinolytic protease from Bacillus circulans strain DZ100 for detergent formulations and feather-biodegradation process. Inter Biodeter Biodegrad 83:129–138
Bouacem K, Bouanane Darenfed A, Laribi-Habchi H, Ben Elhoul M, Hmida Sayari A, Hacene H, Ollivier B, Fardeau ML, Jaouadi B, Bejar S (2015) Biochemical characterization of a detergent-stable serine alkaline protease from Caldicoprobacter guelmensis. Int J Biol Macromol 81:299–307
Daoud L, Hmani H, Ali MB, Jlidi M, Ali MB (2018) An original halo-alkaline protease from Bacillus halodurans strain US193: biochemical characterization and potential use as bio-additive in detergents. J Polym Environ 26:23–32
Doan CT, Tran TN, Nguyen VB, Vo TPK, Nguyen AD, Wang SL (2019) Chitin extraction from shrimp waste by liquid fermentation using an alkaline protease-producing strain, Brevibacillus parabrevis. Int J Biol Macromol 131:706–715
Dorra G, Ines K, Imen BS, Laurent C, Sana A, Olfa T, Pascal C, Thierry J, Ferid L (2018) Purification and characterization of a novel high molecular weight alkaline protease produced by an endophytic Bacillus halotolerans strain CT2. Int J Biol Macromol 111:342–351
Dun Y, Li Y, Xu J, Hu Y, Zhang C, Liang Y, Zhao S (2019) Simultaneous fermentation and hydrolysis to extract chitin from crayfish shell waste. Int J Biol Macromol 123:420–426
Ghorbel Bellaaj O, Younes I, Maâlej H, Hajji S, Nasri M (2012) Chitin extraction from shrimp shell waste using Bacillus bacteria. Int J Biol Macromol 51:1196–1201
Hadjidj R, Badis A, Mechri S, Eddouaouda K, Khelouia L, Annane R, El Hattab M, Jaouadi B (2018) Purification, biochemical, and molecular characterization of novel protease from Bacillus licheniformis strain K7A. Int J Biol Macromol 114:1033–1048
Hamdi M, Hammami A, Hajji S, Jridi M, Nasri M, Nasri R (2017) Chitin extraction from blue crab (Portunus segnis) and shrimp (Penaeus kerathurus) shells using digestive alkaline proteases from P. segnis viscera. Int J Biol Macromol 101:455–463
Hamiche S, Mechri S, Khelouia L, Annane R, El Hattab M, Badis A, Jaouadi B (2019) Purification and biochemical characterization of two keratinases from Bacillus amyloliquefaciens S13 isolated from marine brown alga Zonaria tournefortii with potential keratin-biodegradation and hide-unhairing activities. Int J Biol Macromol 122:758–769
Hammami A, Hamdi M, Abdelhedi O, Jridi M, Nasri M, Bayoudh A (2017) Surfactant- and oxidant-stable alkaline proteases from Bacillus invictae: characterization and potential applications in chitin extraction and as a detergent additive. Int J Biol Macromol 96:272–281
He G, Wang Z, Zheng H, Yin Y, Xiong X, Lin R (2012) Preparation, characterization and properties of aminoethyl chitin hydrogels. Carbohydr Polym 90:1614–1619
Jacobs M, Eliasson M, Uhlen M, Flock JI (1985) Cloning, sequencing and expression of subtilisin Carlsberg from Bacillus licheniformis. Nucleic Acids Res 13:8913–8926
Jain SC, Shinde U, Li Y, Inouye M, Berman HM (1998) The crystal structure of an autoprocessed Ser221Cys-subtilisin E-propeptide complex at 2.0 Å resolution. J Mol Biol 284:137–144
Jellouli K, Ghorbel-Bellaaj O, Ayed HB, Manni L, Agrebi R, Nasri M (2011) Alkaline-protease from Bacillus licheniformis MP1: purification, characterization and potential application as a detergent additive and for shrimp waste deproteinization. Process Biochem 46:1248–1256
Lassoued I, Hajji S, Mhamdi S, Jridi M, Bayoudh A, Barkia A, Nasri M (2015) Digestive alkaline proteases from thornback ray (Raja clavata): characteristics and applications. Int J Biol Macromol 80:668–675
Li Y, Loh YR, Hung AW, Kang C (2018) Characterization of molecular interactions between Zika virus protease and peptides derived from the C-terminus of NS2B. Biochem Biophys Res Commun 503:691–696
Liu S, Sun J, Yu L, Zhang C, Bi J, Zhu F, Qu M, Jiang C, Yang Q (2012) Extraction and characterization of chitin from the beetle Holotrichia parallela motschulsky. Molecules 17:4604–4611
Lopes C, Antelo LT, Franco Uría A, Alonso AA, Pérez Martín R (2018) Chitin production from crustacean biomass: sustainability assessment of chemical and enzymatic processes. J Clean Prod 172:4140–4151
Mao X, Guo N, Sun J, Xue C (2017) Comprehensive utilization of shrimp waste based on biotechnological methods: a review. J Clean Prod 143:814–823
Mechri S, Ben Elhoul Berrouina M, Omrane Benmrad M, Zaraî Jaouadi N, Rekik H, Moujehed E, Chebbi A, Sayadi S, Chamkha M, Bejar S, Jaouadi B (2017a) Characterization of a novel protease from Aeribacillus pallidus strain VP3 with potential biotechnological interest. Int J Biol Macromol 94:221–232
Mechri S, Kriaa M, Ben Elhoul Berrouina M, Omrane Benmrad M, Zaraî Jaouadi N, Rekik H, Bouacem K, Bouanane Darenfed A, Chebbi A, Sayadi S, Chamkha M, Bejar S, Jaouadi B (2017b) Optimized production and characterization of a detergent-stable protease from Lysinibacillus fusiformis C250R. Int J Biol Macromol 101:383–397
Mesbah NM, Wiegel J (2014) Purification and biochemical characterization of halophilic, alkalithermophilic protease AbCP from Alkalibacillus sp. NM-Fa4. J Mol Catal B Enzym 105:74–81
Mohamed S, Bouacem K, Mechri S, Addou NA, Laribi-Habchi H, Fardeau ML, Jaouadi B, Bouanane-Darenfed A, Hacène H (2019) Purification and biochemical characterization of a novel acido-halotolerant and thermostable endochitinase from Melghiribacillus thermohalophilus strain Nari2AT. Carbohydr Res 473:46–56
Mohammed MH, Williams PA, Tverezovskaya O (2013) Extraction of chitin from prawn shells and conversion to low molecular mass chitosan. Food Hydrocoll 31:166–171
Mokashe N, Chaudhari B, Patil U (2018) Operative utility of salt-stable proteases of halophilic and halotolerant bacteria in the biotechnology sector. Int J Biol Macromol 117:493–522
Nirmal NP, Laxman RS (2014) Enhanced thermostability of a fungal alkaline protease by different additives. Enzym Res 2014:109303. https://doi.org/10.1155/2014/109303
Omrane Benmrad M, Moujehed E, Ben Elhoul M, Zaraî Jaouadi N, Mechri S, Rekik H, Kourdali S, El Hattab M, Badis A, Sayadi S, Bejar S, Jaouadi B (2016) A novel organic solvent-and detergent-stable serine alkaline protease from Trametes cingulata strain CTM10101. Int J Biol Macromol 91:961–972
Omrane Benmrad M, Moujehed E, Ben Elhoul M, Mechri S, Bejar S, Zouari R, Baffoun A, Jaouadi B (2018) Production, purification, and biochemical characterization of serine alkaline protease from Penicillium chrysogenium strain X5 used as excellent bio-additive for textile processing. Int J Biol Macromol 119:1002–1016
Peng Y, Yang XJ, Xiao L, Zhang YZ (2004) Cloning and expression of a fibrinolytic enzyme (subtilisin DFE) gene from Bacillus amyloliquefaciens DC-4 in Bacillus subtilis. Res Microbiol 155:167–173
Prameela K, Venkatesh K, Immandi SB, Kasturi APK, Rama Krishna C, Murali Mohan C (2017) Next generation nutraceutical from shrimp waste: the convergence of applications with extraction methods. Food Chem 237:121–132
Purohit MK, Singh SP (2014) Cloning, over expression and functional attributes of serine proteases from Oceanobacillus iheyensis OM A18 and Haloalkaliphilic bacterium OM E12. Process Biochem 49:61–68
Rani K, Rana R, Datt S (2012) Review on latest overview of proteases. Int J Curr Life Sci 2:12–18
Rekik H, Zaraî Jaouadi N, Gargouri F, Bejar W, Frikha F, Jmal N, Bejar S, Jaouadi B (2019) Production, purification and biochemical characterization of a novel detergent-stable serine alkaline protease from Bacillus safensis strain RH12. Int J Biol Macromol 121:1227–1239
Safaie M, Momeni M, Azhdahakoshpour A (2016) The first record of the Speckled shrimp Metapenaeus monoceros (Fabricius, 1798) (Crustacea: decapoda: Penaeidae) from the Iranian coastal waters. Mar Biodivers Rec 9:69
Shakilanishi S, Chandra Babu NK, Shanthi C (2017) Exploration of chrome shaving hydrolysate as substrate for production of dehairing protease by Bacillus cereus VITSN04 for use in cleaner leather production. J Clean Prod 149:797–804
Singh R, Kumar M, Mittal A, Mehta PK (2016) Microbial enzymes: Industrial progress in 21st century. 3 Biotech 6:174
Vojcic L, Pitzler C, Koerfer G, Jakob F, Martinez R, Maurer KH, Schwaneberg U (2015) Advances in protease engineering for laundry detergents. New Biotech 32:629–634
Wang D, Li A, Han H, Liu T, Yang Q (2018) A potent chitinase from Bacillus subtilis for the efficient bioconversion of chitin-containing wastes. Int J Biol Macromol 116:863–868
Yang J, Li J, Mai Z, Tian X, Zhang S (2013) Purification, characterization, and gene cloning of a cold-adapted thermolysin-like protease from Halobacillus sp. SCSIO 20089. J Biosci Bioeng 115:628–632
Younes I, Hajji S, Frachet V, Rinaudo M, Jellouli K, Nasri M (2014) Chitin extraction from shrimp shell using enzymatic treatment. Antitumor, antioxidant and antimicrobial activities of chitosan. Int J Biol Macromol 69:489–498
Younes I, Hajji S, Rinaudo M, Chaabouni M, Jellouli K, Nasri M (2016) Optimization of proteins and minerals removal from shrimp shells to produce highly acetylated chitin. Int J Biol Macromol 84:246–253
Yu P, Huang X, Ren Q, Wang X (2019) Purification and characterization of a H2O2-tolerant alkaline protease from Bacillus sp. ZJ1502, a newly isolated strain from fermented bean curd. Food Chem 274:510–517
Zaraî Jaouadi N, Jaouadi B, Aghajari N, Bejar S (2012) The overexpression of the SAPB of Bacillus pumilus CBS and mutated sapB-L31I/T33S/N99Y alkaline proteases in Bacillus subtilis DB430: new attractive properties for the mutant enzyme. Bioresour Technol 105:142–151
Zaraî Jaouadi N, Rekik H, Badis A, Trabelsi S, Belhoul M, Yahiaoui AB, Ben Aicha H, Toumi A, Bejar S, Jaouadi B (2013) Biochemical and molecular characterization of a serine keratinase from Brevibacillus brevis US575 with promising keratin-biodegradation and hide-dehairing activities. PLoS One 8:e76722
Acknowledgements
The authors would like to express their gratitude to Mr. K. Walha, Mrs. N. Kchaou, and Mrs. N. Masmoudi (Analysis Unit-CBS) for their technical assistance. We would also like to thank Dr. W. Saibi and Pr. H. Belghith (CBS) and Mr. F. Allala (LCBM, FSB-USTHB) for their constructive discussions and suggestions. Special thanks are also due to Pr. W. Hariz from the English Department at the Faculty of Sciences of Sfax (FSS), University of Sfax (Tunisia) for constructive proofreading and language polishing services.
Funding
This study was supported by the Tunisian Ministry of Higher Education and Scientific Research under the Contract Program LMBEE-CBS/code: LR15CBS06_2015-2019 and the Ph.D. Student Fellowship of the Doctoral Institute of Fundamental Sciences of the Sfax University represented by the FSS, University of Sfax/Code: ED08FSSf01.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by M. Moracci.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mechri, S., Bouacem, K., Jabeur, F. et al. Purification and biochemical characterization of a novel thermostable and halotolerant subtilisin SAPN, a serine protease from Melghiribacillus thermohalophilus Nari2AT for chitin extraction from crab and shrimp shell by-products. Extremophiles 23, 529–547 (2019). https://doi.org/10.1007/s00792-019-01105-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-019-01105-8