
Abstract
Caldicellulosiruptor bescii, the most thermophilic cellulolytic bacterium, is rich in hydrolytic and accessory enzymes that can degrade untreated biomass, but the precise role of many these enzymes is unknown. One of such enzymes is a predicted GDSL lipase or esterase encoded by the locus Athe_0553. In this study, this probable esterase named as Cbes-AcXE2 was overexpressed in Escherichia coli. The Ni–NTA affinity purified enzyme exhibited an optimum pH of 7.5 at an optimum temperature of 70 °C. Cbes-AcXE2 hydrolyzed p-nitrophenyl (pNP) acetate, pNP-butyrate, and phenyl acetate with approximately equal efficiency. The specific activity and K M for the most preferred substrate, phenyl acetate, were 142 U/mg and 0.85 mM, respectively. Cbes-AcXE2 removed the acetyl group of xylobiose hexaacetate and glucose pentaacetate like an acetyl xylan esterase (AcXE). Bioinformatics analyses suggested that Cbes-AcXE2, which carries an SGNH hydrolase-type esterase domain, is a member of an unclassified carbohydrate esterase (CE) family. Moreover, Cbes-AcXE2 is evolutionarily and biochemically similar to an unclassified AcXE, Axe2, of Geobacillus stearothermophilus. Thus, we proposed a novel family of carbohydrate esterase for both Cbes-AcXE2 and Axe2.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lignocellulosic biomass is a renewable feedstock used to produce biofuels and green chemicals and to reduce fossil fuel dependence (Ellabban et al. 2014; Kerckhoffs and Renquist 2013; Kopetz 2013). Numerous cellulolytic microbes have been harnessed in biomass refineries for biomass hydrolysis and transformation. However, to date, achieving a cost-effective enzymatic release of fermentable sugars from complex and recalcitrant biomass remains a challenge (Fatih Demirbas 2009; Hasunuma et al. 2013; Himmel et al. 2007). In biomass hydrolysis, these microbes either act as sources of enzyme mixtures that deconstruct the biomass or they produce fuels and chemicals directly from the biomass in a single step of consolidated bioprocessing (Fatih Demirbas 2009; Himmel et al. 2007).
Thermophilic cellulolytic microbes have been regarded as a potential inexpensive alternative because of their advantages associated with high temperature. A higher reaction rate and substrate loading, the minimum risk of contamination, and the ease of product recovery at a higher temperature render the overall process highly cost-effective (Blumer-Schuette et al. 2008, 2014). Numerous thermophilic genera, namely Clostridia, Caldicellulosiruptor, Acidothermus, Geobacillus, and Thermobifida, have been reported to degrade cellulose or complex biomass (Blumer-Schuette et al. 2014). Among these microbes, Caldicellulosiruptor bescii has the highest optimum growth temperature of 78 °C and can grow up to 90 °C (Yang et al. 2010). In addition, this bacterium utilizes various insoluble polymeric substrates as carbon sources, such as raw switchgrass, crystalline cellulose, xylan, pectin, and starch (Basen et al. 2014; Yang et al. 2010). Therefore, efforts are being focused on elucidating the set of unique enzymes involved in the degradation and utilization of these substrates to achieve an improved understanding of the molecular mechanism underlying C. bescii-based biomass hydrolysis.
To date, based on the transcriptomic and proteomic studies on C. bescii, the functional roles of only a few cellulolytic, xylanolytic, and pectinolytic enzymes have been identified (Dam et al. 2011; Kataeva et al. 2013; Lochner et al. 2011). Of these characterized enzyme groups, a secreted multidomain cellulase, CelA (Athe_1857), has been reported to be primarily involved in the solubilization of crystalline cellulose (Brunecky et al. 2013; Dam et al. 2011). Deletion of the corresponding CelA-encoding gene from the genome drastically diminishes cell’s ability to grow on cellulose and switchgrass (Young et al. 2014). Similarly, Su et al. identified and explored xylan degradation-related enzymes. An operon of six genes (Athe_182–187) encoding two endoxylanases, two xylosidases, a probable acetylxylanesterase (AcXE), and a glycoside hydrolase family 43 domain containing protein was involved in xylan utilization (Su et al. 2013). In addition, recombinant versions of these six xylanolytic enzymes were used to design a thermostable enzyme cocktail for xylan hydrolysis (Su et al. 2013). Furthermore, a pectinolytic enzyme (Athe_1854) was reported to function synergistically with CelA in raw switchgrass degradation (Alahuhta et al. 2013).
Although several studies on C. bescii have been reported, the functions of many enzymes from C. bescii, including many hydrolases and polysaccharide lyases associated with biomass degradation, remain undetermined. The present study focuses on the probable AcXE encoded by the locus Athe_0553, which was found to be upregulated during growth on switchgrass, but its function was not established in the previous study by Kataeva et al. (2013).
AcXEs are essential for the removal of acetyl groups attached to xylopyranosyl, glucuronoxylan, and galactoglucomannan of the heterogeneous hemicellulose. This deacetylation reaction is crucial for overall biomass hydrolysis, because hemicellulose constitutes approximately 20–30% of the total lignocellulosic material (Biely 2012; Biely et al. 1985; Mackenzie et al. 1987; Pawar et al. 2013; Zhang et al. 2011). AcXEs are either extracellular or intracellular and remove the acetyl groups located at the O-2 or O-3 positions of xylopyranosyl residues. Typically, microbes degrade polymeric xylan into soluble deacetylated xylooligosaccharides (XOSs) through a synergistic action of secreted endoxylanase and AcXEs. Subsequently, XOSs are transported inside the cell, where β-xylosidases produce fermentable xylose sugars (Biely 2012; Pawar et al. 2013; Su et al. 2013). AcXEs play a fundamental role in the overall xylan utilization, because acetyl group removal is essential for β-xylosidase action (Biely et al. 1996, 2000; Zhang et al. 2011).
To date, only one AcXE1 of C. bescii, which was encoded by the locus Athe_0152 but not upregulated, has been studied in detail (Su et al. 2013). AcXE1 together with endoxylanase and β-xylosidase was reported to increase xylose yield in oat-spelt xylan (OSX) hydrolysis (Su et al. 2013).
Moreover, two probable AcXEs, Athe_0186 and Athe_0553, have been found to be upregulated by 15- and 4-fold, respectively, which validates their specific roles in biomass degradation (Chowdhary et al. 2015; Kataeva et al. 2013). Therefore, the biochemical characterization of these upregulated AcXEs would reveal their specific roles in xylan utilization. Moreover, a previous study was unsuccessful in overexpressing Athe_0186, a member of the six-gene cluster associated with xylan degradation, in Escherichia coli (Su et al. 2013). In the present study, another enzyme, predicted as a GDSL lipase and named as Cbes-AcXE2, was overexpressed in E. coli. The purified protein was characterized in detail. Our study suggests that Cbes-AcXE2is a member of an unclassified thermostable AcXE that acts on soluble acetylated xylobiose and glucose.
Materials and methods
Gene cloning and protein expression
Cbes-AcXE2 [National Center for Biotechnology Information (NCBI) Reference Sequence ACM59679] was polymerase chain reaction (PCR)-amplified from the genome of C. bescii (DSM 6725). The PCR was performed using PrimeSTAR HS DNA polymerase premix (DSS Takara Bio India Pvt. Ltd., New Delhi, India) and a set of primers. The following primers were used: (1) forward primer, 5′-GACGGGTGTGCTAGCAAGATTGAAAATGGAAGCAAACTTCTTTTCATAG-3′ and (2) reverse primer, 5′-GAGAGTGGCTCTCGAGAAACTCTATAGCTTTTAAAAACGCATTGG-3′ (the NheI and XhoI restriction sites are represented in italics). The amplified product was cloned into the pET-23a plasmid at the NheI and XhoI restriction sites, and this recombinant plasmid was transformed into the E. coli BL21(DE3) cells for protein overexpression. The protein was expressed by growing the E. coli BL21(DE3) cells at 37 °C, with constant shaking at 200 rpm for 24 h in an autoinduction terrific growth medium with trace elements (Hi-Media Laboratories Pvt. Ltd., Mumbai, India).
Protein purification, quantification, and electrophoresis
The grown cells were resuspended in a lysis buffer (20 mM Tris, pH 8.2) and lysed using the freeze–thaw method. Subsequently, the cell lysate was heated at 70 °C for 30 min, and the cell-free extract was prepared by centrifuging the heated cell lysate at 9500 rpm for 40 min. The supernatant was loaded onto a lysis buffer pre-equilibrated 1.5 ml Ni–NTA agarose column (Qiagen, Venlo, The Netherlands). Subsequently, the bound proteins were eluted with an imidazole-containing elution buffer (1 M imidazole; 20 mM Tris, pH 8.2) and 1% glycerol. Protein estimation of the eluted fractions was performed using the Bradford method, and the purified protein was analyzed through 12.0% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) to assess the purity and molecular weight according to the Laemmli SDS-PAGE protocol (Bradford 1976; Laemmli 1970).
Enzyme assay
The purified Cbes-AcXE2 was concentrated and buffer-exchanged in a 3-kDa centrifugal concentrator (Pall Corporation, India). The protein was quantified by measuring UV absorbance at 280 nm based on the molar extinction coefficient (34.045 mM−1 cm−1). Furthermore, p-nitrophenyl (pNP)-acetate, pNP-butyrate, pNP-octanoate, pNP-dodeconate, phenyl acetate, N-acetylglucosamine, glucose pentaacetate, OSX, except 1,4-β-xylobiosehexacetate and birchwood xylan were used in the assays. All these substrates were purchased from Sigma-Aldrich (St Louis, USA) except 1,4-β-xylobiosehexacetate, which was procured from Carbosynth (UK). The acetylation of OSX was performed on the basis of a previously reported protocol, and acetylation was confirmed through the gravimetric analysis of acetylated OSX (Biely et al. 1996).
A 3-ml standard assay mixture comprised approximately 1 µg of Cbes-AcXE2 and 0.375 mM pNP esters. The released pNP after hydrolysis was continuously measured at 410 nm using a UV-2550 UV–Vis spectrophotometer (Shimadzu, Kyoto, Japan) coupled with an accessory TCC-240A for cuvette temperature control. The standard enzyme assays were performed in the following buffers: 100 mM citrate (pH 5.0–6.0), 100 mM phosphate (pH 7.0–7.5), and 100 mM Tris (pH 8.0–9.0). The molar extinction coefficients of pNP at 410 nm were 1.8, 9.6, 13.3, 17.4, and 18.1 mM−1 cm−1 at pH 6.0, 7.0, 7.5, 8.0, and 9.0, respectively. Similarly, at pH 7.5, the same standard enzyme assays were performed in a temperature range of 40–90 °C to determine the optimum conditions of Cbes-AcXE2. One unit of enzyme corresponds to the release of 1 µM pNP/min/ml.
In the phenyl acetate assay, 0.21-M substrate (Sigma-Aldrich, St Louis, USA) was used with approximately 12 µg of the purified protein in a total reaction volume of 3 ml. The phenol released after hydrolysis was progressively measured at 270 nm. The molar extinction coefficient of phenol was 1.67 mM−1 cm−1 at 270 nm in a 100-mM phosphate buffer of pH 7.5.
The enzyme activity with acetylated substrates was determined by quantifying the release of acetic acid from substrates. Approximately 1 mg of N-acetylglucosamine, glucose pentaacetate, 1,4-β-xylobiose hexaacetate, OSX, acetylated OSX, and birchwood xylan were incubated with 1–2 µg of the protein in an assay mixture of 400 µl. Acetic acid release was measured using the high-performance liquid chromatography (HPLC)-based method as described previously (Mackenzie et al. 1987). An Agilent HPLC model 1260 equipped with an Aminex 87 H column (300 × 7.8 mm) and a variable wavelength detector was used to measure acetic acid release at 210 nm. The column was operated at 65 °C, with a flow rate of 0.6 ml/min and 5-mM sulfuric acid as an eluent.
Enzyme kinetics
The kinetic parameters, namely K M, V max, and catalytic efficiency, were determined from the Lineweaver–Burk plot for the assays performed in 100-mM sodium phosphate buffer (pH 7.5) at 70 °C. In the two assays, pNP-acetate (0.01–5.0 mM) and phenyl acetate (0.1–2.0 mM) were used as substrates, and 1 µg of Cbes-AcXE2 was used in both assays. The enzymatic reactions were monitored through UV–Vis spectroscopy as described in the previous section. The initial velocities of both enzymatic reactions were plotted in the Lineweaver–Burk double-reciprocal plot to estimate the kinetic parameters.
Thermostability and fluorescence intensity measurements
The thermal stability of the protein was determined by incubating the protein in a buffer (pH 7.5) at 70 °C, and enzyme activity was measured at different time points by performing standard assays with pNP-acetate. Furthermore, the same protein sample was used to collect the fluorescence emission data at an excitation wavelength of 280 nm and a band width of 5 nm on a Jasco FP-6500 spectrofluorometer. The collected data represent an average of 10 scans.
Dynamic light scattering
The purified and filtered protein solution (0.684 mg/ml) in 50-mM phosphate buffer (pH 7.2) was used to measure the hydrodynamic radius on a Möbiuζ light scattering instrument (Wyatt Technology Corporation, USA). The data were acquired for every 5 s, with 50 acquisitions for five independent runs. The collected data were analyzed using Dynamics software to calculate the average radius, diffusion coefficient, and polydispersity.
Bioinformatics analyses and homology modeling
NCBI protein Basic Local Alignment Search Tool (BLAST) was performed using the online BLAST web interface (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins) (Johnson et al. 2008). The protein sequences for different carbohydrate esterase (CE) families were retrieved from the Universal Protein Resource Knowledgebase. The phylogenetic studies of multiple aligned protein sequences were performed through Molecular Evolutionary Genetics Analysis (MEGA) 5.05 using the default parameters (Tamura et al. 2007). A phylogenetic tree was constructed on the basis of the neighbor-joining statistical method and the poisson model of amino-acid substitution with bootstrap replications of 1000. The sequence alignment was performed using Clustal Omega, and the predicted secondary structure was formatted and demonstrated using the ESPript3 online server (Robert and Gouet 2014; Sievers et al. 2011).
Homology modeling created a structural model of Cbes-AcXE2 on the template of Axe2 from Geobacillus stearothermophilus (PDB ID: 3W7V). The default parameters of the Swiss Model (http://swissmodel.expasy.org/) server were used to predict the structural model (Biasini et al. 2014). The graphics and three-dimensional structure were represented using a molecular visualization tool, Pymol (The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC).
Results and discussion
Cbes-AcXE2 is a novel SGNH hydrolase-type esterase domain containing AcXE
The Carbohydrate-Active Enzyme database has grouped esterases working on carbohydrates into 16 CE families (Lombard et al. 2014). AcXEs (EC 3.1.1.72) acting on acetylated xylan have been distributed into eight CE families, CE1-7 and 16 (Biely 2012; Pawar et al. 2013). Although the CE12 family has two probable AcXEs, the deacetylation activity of both enzymes on xylan is yet to be established (Adesioye et al. 2016). AcXEs present in the eight CEs, except CE4, have the standard catalytic triad, Ser-His-Asp (Glu), of the serine esterase in a conserved motif of either GxSxG or GDSL (Akoh et al. 2004). Among these CEs, CE3, CE6, and CE16 also have an SGNH hydrolase-type esterase domain (Ašler et al. 2010). Furthermore, the SGNH hydrolase domain is present in viral hemagglutinin-esterase surface glycoproteins, a mammalian acetylhydrolase, a rhamnogalacturonan acetylesterase, and a multifunctional enzyme thioesterase I (TAP) from E. coli (Bielen et al. 2009; Lo et al. 2003; Sánchez et al. 2012).
NCBI Protein BLAST of Cbes-AcXE2 against the RefSeq protein database revealed a conserved domain of the SGNH hydrolase superfamily and observed similar sequences in all known members of Caldicellulosiruptor (all protein sequences are included in online resource 2). As expected, the identical sequences from the same genus had a sequence identity of >90%. Cbes-AcXE2 was found to be upregulated during growth on switchgrass; therefore, the presence of highly identical sequences in all known members of Caldicellulosiruptor may suggest that Cbes-AcXE2 plays an important role in biomass degradation.
Furthermore, multiple sequence alignments of all protein sequences having greater than 40% sequence identity with Cbes-AcXE2 revealed conserved amino-acid residues, namely, S, G, N, and H of the typical SGNH hydrolase superfamily (Fig. 1a). From these protein sequences, only Axe2 from G. stearothermophilus was active on acetylated xylan (Alalouf et al. 2011). Furthermore, Cbes-AcXE2 shared significant sequence identity (58.69%) with Axe2. Based on the sequence alignment, the likely catalytic triad residues in Cbes-AcXE2 were Ser-15, Asp-192, and His-195 (Fig. 1a). The predicted catalytic residues of Cbes-AcXE2 were analyzed further in a three-dimensional structural model. Nonetheless, Axe2 has not been linked to any known CE family; therefore, Cbes-AcXE2 is an unclassified thermostable AcXE with a distinct SGNH hydrolase esterase domain.

a Multiple Sequence alignment of Cbes-AcXE2 with the protein sequences having greater than 40% sequence identity to show the four conserved blocks marked with amino-acid positions number on top row. The conserved residues—S, G, N, and H—in the blocks are labeled with roman numerals with an arrow mark (bottom row). Multiple sequence alignment was performed by Clustal Omega online server and formatted by ESPript 3 online server. b Unrooted neighbor-joining phylogenetic tree of Cbes-AcXE2, Axe2, and identical sequences from Caldicellulosiruptor genus with other members of carbohydrate esterases having SGNH hydroloase esterase domain-CE3, CE6, and CE16. Phylogenetic tree was constructed by MEGA 5.05 software
Using the neighbor-joining phylogenetic tree method, Cbes-AcXE2, Axe2, and identical sequences from Caldicellulosiruptor were analyzed together with three CEs having a SGNH hydrolase-type esterase domain. Cbes-AcXE2 was not related to any known CEs with SGNH hydrolase-type esterase domain (Fig. 1b). Notably, Cbes-AcXE2 was grouped with Axe2 from G. stearothermophilus, which was previously proposed as a novel AcXE family (Alalouf et al. 2011). All other homologous protein sequences were also grouped together with Cbes-AcXE2 and Axe2. Furthermore, Cbes-AcXE2 and Axe2 differed in terms of their sequence identity and phylogeny with all three CEs, namely CE3, CE6, and CE16, having SGNH hydrolase esterase domain.
Overexpression and purification of recombinant Cbes-AcXE2
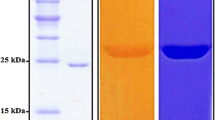
Cbes-AcXE2 was overexpressed in E. coli BL21 (DE3) cells using a T7 promoter-based expression system, and the protein was obtained in the soluble fraction of the cell lysate. The C-terminus 6X-Histag was used to purify the protein through Ni–NTA affinity chromatography. The overall protein yield was approximately 4–5 mg/g wet cell weight. SDS-PAGE of the eluted fractions (Fig. 2a) revealed that the purified protein has a molecular weight of approximately 25 kDa, which is consistent with the predicted molecular weight of 25.8 kDa.

a SDS-PAGE profile of the eluted protein from Ni–NTA affinity chromatography with protein molecular weight marker in the left lane. b, c Relative activity at different pH and temperatures, respectively. All reactions were incubated for 30 min and performed in triplicate. For different pH, the incubation temperature was 70 °C, and for different temperature, pH 7.5 was used in the assays. d Relative activity with different substrates; pNP C-2 (acetate); pNP C-4 (butyrate); pNP C-8 (octanoate); pNP C-12 (dodecanoate); and PA (phenyl acetate). All assays were performed in triplicates
Temperature and pH optima of Cbes-AcXE2
pNP-acetate was used to determine the optimum temperature and pH of the enzyme. The optimum pH and temperature were 7.5 and 70 °C, respectively (Fig. 2b, c). The optimum conditions of Cbes-AcXE2 were very close to the optimum growth conditions of C. bescii (approximately pH 7.0 and 78 °C). However, the temperature optimum of AcXE2 was 8 °C lower than the optimum growth temperature of C. bescii (Yang et al. 2010). A similar difference in the optimum temperature has also been observed in the recombinant version of CelA, which lacked glycosylation compared with the native CelA (Brunecky et al. 2013; Chung et al. 2015). Therefore, posttranslational modifications could play a role in lowering the temperature optimum of Cbes-AcXE2.
Substrate specificity of Cbes-AcXE2
Under optimum temperature and pH conditions, the enzyme hydrolyzed pNP-butyrate and pNP-acetate with almost similar efficiency. However, only approximately 10–15% relative activity of the enzyme was observed with pNP-octanoate compared with pNP-acetate (Fig. 2d). In addition, Cbes-AcXE2 was active on phenyl acetate, a standard substrate of AcXE. The specific enzyme activity with phenyl acetate was 142 mM min−1 mg−1, which was significantly higher than that with pNP-acetate (Table 1; Fig. 2d). A higher specific activity with phenyl acetate is common among AcXEs. Furthermore, AcXEs have been reported to prefer pNP-ester-containing short-chain fatty acids (Biely 2012; Biely et al. 1985, 1996).
To investigate whether the purified protein can deacetylate the naturally acetylated substrate, we evaluated polymeric OSX, birchwood xylan, and acetylated OSX. However, Cbes-AcXE2 could not deacetylate any tested polymeric acetylated xylan (data not shown). By contrast, the evolutionary related protein, Axe2 from G. stearothermophilus hydrolyzed 20–30% acetyl groups from completely acetylated birchwood xylan (Alalouf et al. 2011).
In addition, soluble acetylated substrates, namely glucose pentaacetate and 1,4-β-xylobiose hexaacetate, were analyzed to explore deacetylation activity of the enzyme. One microgram of Cbes-AcXE2 removed 100% acetyl groups from glucose pentaacetate within 1 h (online resource 1, Fig. 3a). Under similar conditions, only 70–80% acetyl groups were released from 1,4-β-xylobiose hexaacetate (online resource 1, Fig. 3a). Here, when the enzyme dosage was increased to 2 µg, 1,4-β-xylobiose hexaacetate was also completely hydrolyzed in 1 h (online resource 1). In our experiments, the slower deacetylation rate of the enzyme on acetylated xylobiose than on glucose pentaacetate may be attributed to the dimeric nature of 1,4-β-xylobiose hexaacetate. Although N-acetylglucosamine was also used in the assays, the enzyme could not deacetylate the N-linked acetyl groups (Fig. 3a). Altogether, these results confirmed that Cbes-AcXE2 is an O-linked deacetylase, which acts only on the O-linked acetyl groups of glucose and xylobiose but not on polymeric insoluble xylan.

a Kinetic data for acetic acid release from acetylated substrates (labeled in top panel). The assays were done in triplicates at 70 °C in a 100-mM phosphate buffer of pH 7.5. The acetic acid was quantified by Aminex 87 H column on an HPLC using variable wavelength detector at 210 nm. The standard acetic acid graph is shown in online resource 1 (Fig. 1a). b Thermal inactivation kinetics of Cbes-AcXE2. The protein was incubated for 0, 30, 60, 90, 120, 180, 240, and 300 min in triplicates at 70 °C in a 100-mM phosphate buffer of pH 7.5 followed by an assay with pNP-acetate under similar conditions for 30 min
In this study, we have not analyzed the reaction intermediates of the complete deacetylation reaction of fully acetylated glucose and xylobiose. Therefore, it is likely that the enzyme does not have any positional specificity toward the substrates. In contrast, AcXEs exhibit positional specificity, because acetylated glucuronoxylan present in the hardwood is mostly monoacetylated at position 2 or 3 or 2,3-di-O-acetylated (Adesioye et al. 2016; Biely et al. 2016). Therefore, further nuclear magnetic resonance and thin layer chromatography studies are warranted to investigate the intermediates of the aforementioned reactions and to establish the accurate role and positional specificity of Cbes-AcXE2 in the degradation of polymeric xylan-derived acetylated XOSs.
Evidently, Cbes-AcXE2 has a distinct substrate specificity compared with the SGNH hydrolase-type esterase domain containing three CEs, namely CE3, CE6, and CE16. Cbes-AcXE2 prefers acetylated XOSs as substrates; however, a thermostable CE3 family AcXE, Ces3, from Clostridium thermocellum was highly active on polymeric acetylated xylan (Correia et al. 2008). Similarly, members of the CE6 and CE16 families have broad substrate specificity for polymeric acetylated xylan as well as soluble acetylated XOSs (Adesioye et al. 2016; Alalouf et al. 2011).
Our study finding suggests that Cbes-AcXE2 can be an accessory enzyme in the xylanolytic enzyme inventory, which has not been reported previously. Presumably, in C. bescii-based xylan utilization, Cbes-AcXE2 would participate in the deacetylation of transported extracellular XOSs, which are produced by the action of secreted endoxylanases. Therefore, it is likely that in C. bescii-based xylan utilization, the secreted endoxylanases will generate acetylated XOSs, which are subsequently transported inside the cell for degradation by intracellular Cbes-AcXE2.
Thermostability of Cbes-AcXE2
The thermostability of Cbes-AcXE2 was evaluated at 70 °C for 3 h by assessing pNP-acetate hydrolysis. The protein was stable for approximately 1.5 h, without any significant activity loss (Fig. 3b). Notably, the protein was thermally activated in the first hour of incubation. However, any structural adjustments in the recombinant Cbes-AcXE2 were very unlikely because the protein had already been exposed to similar temperature conditions as the purification step. Therefore, to further investigate the aforementioned observation, the protein samples used for the thermostability measurements were also evaluated to assess the changes in the intrinsic fluorescence intensity of the protein. Notably, a significant increase in the fluorescence intensity along with a small change in the emission maxima (λ max) was observed during the first hour of incubation (online resource 3). Therefore, it is plausible that the structural elements of the protein were more stably folded during the first hour of incubation at 70 °C. Moreover, additional structural investigations of the molecular mechanisms governing the thermostability of Cbes-AcXE2 will provide insights about the thermal adaptability of this protein.
Catalytic efficiency of Cbes-AcXE2
The Michaelis–Menten constants of Cbes-AcXE2 were calculated by plotting the initial velocities of Cbes-AcXE2 with respect to pNP-acetate and phenyl acetate in the Lineweaver–Burk plot. K M, k cat, V max, and catalytic efficiency were determined using both substrates (Fig. 4a, b; Table 1). The K M values were approximately same for both substrates, but the catalytic efficiency of pNP-acetate was twofold more than that of phenyl acetate. In comparison of Axe2, Cbes-AcXE2 was more efficient (Table 1). Notably, Cbes-AXE2 had a 12- and 20-fold lower K M for phenyl acetate and pNP-acetate, respectively (Alalouf et al. 2011). Apparently, this lower K M, which indicates increased substrate affinity, could be attributed to the superiority of Cbes-AcXE2 over Axe2.

a, b Lineweaver–Burk plot of the initial velocity data obtained for pNP-acetate and phenyl acetate, respectively. The assays were done in triplicates at 70 °C in a 100-mM phosphate buffer of pH 7.5. The linear fit and its equation are shown at the top left corner. Based on this, the Michaelis–Menten constants were calculated and shown in Table 1
Structural model of Cbes-AcXE2
A reliable three-dimensional structural model for Cbes-AcXE2 was constructed using the template of Axe2 from G. stearothermophilus (PDB ID: 3W7V) (Lansky et al. 2014). The model had a global mean quality estimate score of 0.82, indicating an accurate predicted three-dimensional structure. The model superimposed satisfactorily with the evolutionary related protein Axe2, with a root-mean-square deviation of 0.217 Å. Figure 5a shows the predicted model with overlaid catalytic triad residues and the loop involved in catalysis. The positions of Ser, His, and Asp (catalytic triad) were similar to the predicted residue positions of 15, 192, and 195, respectively, as described in the previous sections. Remarkably, the orientation of the loop around the active site was significantly altered (Fig. 5a), which could be a major factor leading to the differences between the catalytic efficiency and K M values of Cbes-AcXE2 and Axe2.

a Three-dimensional structural model of Cbes-AcXE2 shown in green color. The active site residues and a loop of the model (red color) were superimposed to the Axe2 of G. stearothermophilus (PDB ID 3W7V) (cyan color). Catalytic residues—Ser 15 Asp 192 and His 195—are shown in ball-stick. b Geometry of catalytic triad residues (red color) of Cbes-AcXE2—Ser 15 Asp 192 and His 195. Residue position and the distance between these residues are also shown
The geometry of the residues forming the catalytic triad was analyzed (Fig. 5b). The catalytic Serine was positioned at a distance of 2.9 Å, and Asp was located 3.8 Å away from His. These observations suggested that the catalytic triad residues are apparently located at an optimum distance to facilitate enzyme catalysis (Hedstrom 2002; Kraut 1977; Radisky et al. 2006).
The previous X-ray crystallography studies have shown that Axe2 has an octameric doughnut-shaped assembly, in which an asymmetric unit of the dimer is assembled into an octamer through salt bridges and hydrogen bonds (Lansky et al. 2013, 2014). Therefore, we determined the absolute molecular weight of Cbes-AcXE2 through dynamic light scattering measurements. The molecular weight of Cbes-AcXE2 was approximately 185.6 kDa with a hydrodynamic radius of 5.5 nm (Fig. 6), which demonstrated a compact octamer of the monomeric 25.8-kDa Cbes-AcXE2. Therefore, Cbes-AcXE2 and Axe2 not only shared a similar substrate specificity but also an exclusive quaternary structure.

Processed dynamic light scattering data of Cbes-AcXE2 (0.684 mg/ml) in 50-mM phosphate buffer of pH 7.5. The data are average of 50 different acquisitions of 5 different runs. The graph shows hydrodynamic radius and the table shows the polydispersity, diffusion coefficient, and the absolute molecular weight
Furthermore, the geometric positions of the crucial residues involved in salt bridges and hydrogen bonds were reported to be responsible for the octameric assembly of Axe2 (Lansky et al. 2014). A structural comparison between these residues indicated that the residues for tetramer and octamer formation were well conserved. However, the residues involved in the assembly of a monomer into a dimer of Cbes-AcXE2 (data not shown) were not conserved. Therefore, we speculated that a monomer is assembled into dimeric Cbes-AcXE2 through a different mechanism. Further X-ray crystallography studies on the monomer–dimer assembly are warranted to determine the orientation of the active site in the multimeric Cbes-AcXE2.
Conclusions
In the current study, we performed the biochemical characterization of an AcXE of the most thermophilic cellulolytic bacterium, C. bescii. Our results demonstrated that Cbes-AcXE2 acts on acetylated xylobiose and glucose. Furthermore, Cbes-AcXE2 was upregulated during growth on switchgrass, and all known members of Caldicellulosiruptor had very similar protein sequences. Altogether, we suggest that Cbes-AcXE2 plays a pivotal role in plant biomass degradation. In addition, Cbes-AcXE2 shares sequence identity, substrate specificity, and a unique octameric assembly with Axe2, which has not been included in any CEs. Therefore, we propose a novel CE family, CE17, for Cbes-AcXE2 as well as for the previously studied Axe2. These suggestions are consistent with those of a study by Adesioye et al. (2016).
References
Adesioye FA, Makhalanyane TP, Biely P, Cowan DA (2016) Phylogeny, classification and metagenomic bioprospecting of microbial acetyl xylan esterases. Enzyme Microb Technol 93–94:79–91. doi:10.1016/j.enzmictec.2016.07.001
Akoh CC, Lee GC, Liaw YC et al (2004) GDSL family of serine esterases/lipases. Prog Lipid Res 43:534–552. doi:10.1016/j.plipres.2004.09.002
Alahuhta M, Brunecky R, Chandrayan P et al (2013) The structure and mode of action of Caldicellulosiruptor bescii family 3 pectate lyase in biomass deconstruction. Acta Crystallogr Sect D Biol Crystallogr 69:534–539. doi:10.1107/S0907444912050512
Alalouf O, Balazs Y, Volkinshtein M et al (2011) A new family of carbohydrate esterases is represented by a GDSL hydrolase/acetylxylan esterase from Geobacillus stearothermophilus. J Biol Chem 286:41993–42001. doi:10.1074/jbc.M111.301051
Ašler IL, Ivić N, Kovačić F et al (2010) Probing enzyme promiscuity of SGNH hydrolases. Chembiochem 11:2158–2167. doi:10.1002/cbic.201000398
Basen M, Rhaesa AM, Kataeva I et al (2014) Degradation of high loads of crystalline cellulose and of unpretreated plant biomass by the thermophilic bacterium Caldicellulosiruptor bescii. Bioresour Technol 152:384–392. doi:10.1016/j.biortech.2013.11.024
Biasini M, Bienert S, Waterhouse A et al (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. doi:10.1093/nar/gku340
Bielen A, Ćetković H, Long PF et al (2009) The SGNH-hydrolase of Streptomyces coelicolor has (aryl) esterase and a true lipase activity. Biochimie 91:390–400. doi:10.1016/j.biochi.2008.10.018
Biely P (2012) Microbial carbohydrate esterases deacetylating plant polysaccharides. Biotechnol Adv 30:1575–1588. doi:10.1016/j.biotechadv.2012.04.010
Biely P, Puls J, Schneider H (1985) Acetyl xylan esterases in fungal cellulolytic systems. FEBS Lett 186:80–84. doi:10.1016/0014-5793(85)81343-0
Biely P, Côté GL, Kremnický L et al (1996) Substrate specificity of acetylxylan esterase from Schizophyllum commune: mode of action on acetylated carbohydrates. Biochim Biophys Acta Protein Struct Mol Enzymol 1298:209–222. doi:10.1016/S0167-4838(96)00132-X
Biely P, Hirsch J, la Grange DC et al (2000) A chromogenic substrate for a beta-xylosidase-coupled assay of alpha-glucuronidase. Anal Biochem 286:289–294. doi:10.1006/abio.2000.4810
Biely P, Singh S, Puchart V (2016) Towards enzymatic breakdown of complex plant xylan structures: state of the art. Biotechnol Adv 34:1260–1274. doi:10.1016/j.biotechadv.2016.09.001
Blumer-Schuette SE, Kataeva I, Westpheling J et al (2008) Extremely thermophilic microorganisms for biomass conversion: status and prospects. Curr Opin Biotechnol 19:210–217
Blumer-Schuette SE, Brown SD, Sander KB et al (2014) Thermophilic lignocellulose deconstruction. FEMS Microbiol Rev 38:393–448
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi:10.1016/0003-2697(76)90527-3
Brunecky R, Alahuhta M, Xu Q et al (2013) Revealing nature’s cellulase diversity: the digestion mechanism of Caldicellulosiruptor bescii CelA. Science 342:1513–1516. doi:10.1126/science.1244273
Chowdhary N, Selvaraj A, KrishnaKumaar L, Kumar GR (2015) Genome wide re-annotation of Caldicellulosiruptor saccharolyticus with new insights into genes involved in biomass degradation and hydrogen production. PLoS One 10:e0133183. doi:10.1371/journal.pone.0133183
Chung D, Young J, Bomble YJ et al (2015) Homologous expression of the Caldicellulosiruptor bescii cela reveals that the extracellular protein is glycosylated. PLoS One. doi:10.1371/journal.pone.0119508
Correia MAS, Prates JAM, Brás J et al (2008) Crystal structure of a cellulosomal family 3 carbohydrate esterase from Clostridium thermocellum provides insights into the mechanism of substrate recognition. J Mol Biol 379:64–72. doi:10.1016/j.jmb.2008.03.037
Dam P, Kataeva I, Yang SJ et al (2011) Insights into plant biomass conversion from the genome of the anaerobic thermophilic bacterium Caldicellulosiruptor bescii DSM 6725. Nucleic Acids Res 39:3240–3254. doi:10.1093/nar/gkq1281
Ellabban O, Abu-Rub H, Blaabjerg F (2014) Renewable energy resources: current status, future prospects and their enabling technology. Renew Sustain Energy Rev 39:748–764
Fatih Demirbas M (2009) Biorefineries for biofuel upgrading: a critical review. Appl Energy. doi:10.1016/j.apenergy.2009.04.043
Hasunuma T, Okazaki F, Okai N et al (2013) A review of enzymes and microbes for lignocellulosic biorefinery and the possibility of their application to consolidated bioprocessing technology. Bioresour Technol 135:513–522. doi:10.1016/j.biortech.2012.10.047
Hedstrom L (2002) Serine protease mechanism and specificity. Chem Rev 102:4501–4523. doi:10.1021/cr000033x
Himmel ME, Ding S-Y, Johnson DK et al (2007) Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science 315:804–807. doi:10.1126/science.1137016
Johnson M, Zaretskaya I, Raytselis Y et al (2008) NCBI BLAST: a better web interface. Nucleic Acids Res. doi:10.1093/nar/gkn201
Kataeva I, Foston MB, Yang S-J et al (2013) Carbohydrate and lignin are simultaneously solubilized from unpretreated switchgrass by microbial action at high temperature. Energy Environ Sci 6:2186. doi:10.1039/c3ee40932e
Kerckhoffs H, Renquist R (2013) Biofuel from plant biomass. Agron Sustain Dev 33:1–19
Kopetz H (2013) Renewable resources: build a biomass energy market. Nature 494:29–31. doi:10.1038/494029a
Kraut J (1977) Serine proteases: structure and mechanism of catalysis. Annu Rev Biochem 46:331–358. doi:10.1146/annurev.bi.46.070177.001555
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi:10.1038/227680a0
Lansky S, Alalouf O, Solomon V et al (2013) Crystallization and preliminary crystallographic analysis of Axe2, an acetylxylan esterase from Geobacillus stearothermophilus. Acta Crystallogr Sect F Struct Biol Cryst Commun 69:430–434. doi:10.1107/S1744309113004260
Lansky S, Alalouf O, Solomon HV et al (2014) A unique octameric structure of Axe2, an intracellular acetyl-xylooligosaccharide esterase from Geobacillus stearothermophilus. Acta Crystallogr Sect D Biol Crystallogr 70:261–278. doi:10.1107/S139900471302840X
Lo YC, Lin SC, Shaw JF, Liaw YC (2003) Crystal structure of Escherichia coli thioesterase I/protease I/lysophospholipase L1: Consensus sequence blocks constitute the catalytic center of SGNH-hydrolases through a conserved hydrogen bond network. J Mol Biol 330:539–551. doi:10.1016/S0022-2836(03)00637-5
Lochner A, Giannone RJ, Rodriguez M et al (2011) Use of label-free quantitative proteomics to distinguish the secreted cellulolytic systems of Caldicellulosiruptor bescii and Caldicellulosiruptor obsidiansis. Appl Environ Microbiol 77:4042–4054. doi:10.1128/AEM.02811-10
Lombard V, Golaconda Ramulu H, Drula E et al (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. doi:10.1093/nar/gkt1178
Mackenzie CR, Bilous D, Schneider H, Johnson KG (1987) Induction of cellulolytic and xylanolytic enzyme systems in Streptomyces spp. Appl Environ Microbiol 53:2835–2839
Pawar PM-A, Koutaniemi S, Tenkanen M, Mellerowicz EJ (2013) Acetylation of woody lignocellulose: significance and regulation. Front Plant Sci 4:118. doi:10.3389/fpls.2013.00118
Radisky ES, Lee JM, Lu C-JK, Koshland DE (2006) Insights into the serine protease mechanism from atomic resolution structures of trypsin reaction intermediates. Proc Natl Acad Sci USA 103:6835–6840. doi:10.1073/pnas.0601910103
Robert X, Gouet P (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. doi:10.1093/nar/gku316
Sánchez DG, Otero LH, Hernández CM et al (2012) A Pseudomonas aeruginosa PAO1 acetylcholinesterase is encoded by the PA4921 gene and belongs to the SGNH hydrolase family. Microbiol Res 167:317–325. doi:10.1016/j.micres.2011.11.005
Sievers F, Wilm A, Dineen D et al (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi:10.1038/msb.2011.75
Su X, Han Y, Dodd D et al (2013) Reconstitution of a thermostable xylan-degrading enzyme mixture from the bacterium Caldicellulosiruptor bescii. Appl Environ Microbiol 79:1481–1490. doi:10.1128/AEM.03265-12
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599. doi:10.1093/molbev/msm092
Yang SJ, Kataeva I, Wiegel J et al (2010) Classification of “Anaerocellum thermophilum” strain DSM 6725 as Caldicellulosiruptor bescii sp. nov. Int J Syst Evol Microbiol 60:2011–2015
Young J, Chung D, Bomble YJ et al (2014) Deletion of Caldicellulosiruptor bescii CelA reveals its crucial role in the deconstruction of lignocellulosic biomass. Biotechnol Biofuels 7:142. doi:10.1186/s13068-014-0142-6
Zhang J, Siika-Aho M, Tenkanen M, Viikari L (2011) The role of acetyl xylan esterase in the solubilization of xylan and enzymatic hydrolysis of wheat straw and giant reed. Biotechnol Biofuels 4:60. doi:10.1186/1754-6834-4-60
Acknowledgements
We thank Prof. M.W.W Adams (University of Georgia, USA) for the kind gift of the genomic DNA of C. bescii. We also thank Dr. Ajit Tiwari for carefully reading the manuscript and providing his suggestions. This research was financially supported by Department of Biotechnology (Government of India) under the program Energy Bioscience Overseas Fellowship (No. BT/NBDB/22/06/2011) and DBT-ICT Centre for Energy Biosciences – Phase II (BT/EB/ICT-Extension/2012).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by H. Atomi.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Soni, S., Sathe, S.S., Odaneth, A.A. et al. SGNH hydrolase-type esterase domain containing Cbes-AcXE2: a novel and thermostable acetyl xylan esterase from Caldicellulosiruptor bescii . Extremophiles 21, 687–697 (2017). https://doi.org/10.1007/s00792-017-0934-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-017-0934-2