
Abstract
Dihydrofolate (DHF) reductase coded by a plasmid of the extremely halophilic archaeon Haloarcula japonica strain TR-1 (HjDHFR P1) shows moderate halophilicity on enzymatic activity at pH 6.0, although there is no significant effect of NaCl on its secondary structure. To elucidate the salt-activation and -inactivation mechanisms of this enzyme, we investigated the effects of pH and salt concentration, deuterium isotope effect, steady-state kinetics, and rapid-phase ligand-binding kinetics. Enzyme activity was increased eightfold by the addition of 500 mM NaCl at pH 6.0, fourfold by 250 mM at pH 8.0, and became independent of salt concentration at pH 10.0. Full isotope effects observed at pH 10.0 under 0–1000 mM NaCl indicated that the rate of hydride transfer, which was the rate-determining step at the basic pH region, was independent of salt concentration. Conversely, rapid-phase ligand-binding experiments showed that the amplitude of the DHF-binding reaction increased and the tetrahydrofolate (THF)-releasing rate decreased with increasing NaCl concentration. These results suggested that the salt-activation mechanism of HjDHFR P1 is via the population change of the anion-unbound and anion-bound conformers, which are binding-incompetent and -competent conformations for DHF, respectively, while that of salt inactivation is via deceleration of the THF-releasing rate, which is the rate-determining step at the neutral pH region.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Halophilic microorganisms, which are frequently archaea, are isolated from salt lakes, salterns, and sometimes commercially distributed natural salts. Since the intracellular salt concentrations of such halophilic microorganisms are identical to the extracellular conditions to escape hyperosmotic stress, the enzymes they produce have adaptation mechanisms for hypersaline environments (Roesser and Müller 2001). However, the primary and tertiary structures of such halophilic enzymes are almost similar to those of non-halophilic homologs produced by organisms living in normal conditions, and it has been unclear how such halophilic enzymes maintain their function under stressful salt conditions.
Dihydrofolate reductase (DHFR) is a famous model enzyme that catalyzes the reduction of dihydrofolate (DHF) to tetrahydrofolate (THF) using nicotinamide adenine dinucleotide phosphate (NADPH) as a coenzyme. It is a ubiquitous enzyme since its product, THF, is essential for the growth and proliferation of cells. DHFR from Escherichia coli (EcDHFR) has been investigated widely for its crystal and solution structures (Sawaya and Kraut 1997; Osborne et al. 2003), structural stability (Perry et al. 1987; Garvey and Matthews 1989), folding kinetics (Kuwajima et al. 1991; Jennings et al. 1993), and catalytic mechanism (Fierke et al. 1987; Wang et al. 2014). The steady-state enzymatic reaction of EcDHFR contains five elementary steps: two binding steps of NADPH and DHF, hydride transfer from NADPH to DHF, and two releasing steps of NADP+ and THF. In addition, the rate-determining step of enzymatic turnover changes from the THF-releasing step at neutral pH to the hydride-transfer step above pH 8.4 (Fierke et al. 1987). Moreover, the enzymatic activity of EcDHFR decreases gradually with increasing NaCl concentration, and this mechanism is explained by the binding of an inorganic cation near the substrate-binding cleft (Ohmae et al. 2013).
Many studies have also been performed on the adaptation mechanisms of DHFR from various environmental bacteria, for example, the moderate thermophile Geobacillus stearothermophilus (Guo et al. 2014), hyperthermophile Thermotoga maritima (Luk et al. 2014), piezo-psychrophile Moritella profunda (Ohmae et al. 2012; Behiry et al. 2014), and extreme halophile Haloferax volcanii. H. volcanii was isolated from the Dead Sea, and produces two DHFR enzymes, HvDHFR 1 and 2, whose optimal KCl concentrations for enzymatic activity are more than 3.5 M and 500 mM, respectively (Wright et al. 2002). Studies on HvDHFR 1 have been performed on its X-ray crystal structure (Pieper et al. 1998), solution structure using nuclear magnetic resonance (NMR) (Binbuga et al. 2007; Boroujerdi and Young 2009), structural stability (Wright et al. 2002), folding kinetics (Gloss et al. 2008), and enzymatic function (Zusman et al. 1989; Ortenberg et al. 2000). Results of these investigations suggested that destabilization of the unfolded state predominantly invokes the salt-induced stabilization and activation of HvDHFR 1. However, studies on the moderately halophilic enzyme HvDHFR 2 are limited (Ortenberg et al. 2000; Wright et al. 2002), and the mechanisms underlying the optimal salt concentration for its enzymatic activity are still unclear.
Previously, we characterized another moderately halophilic DHFR from a plasmid gene of Haloarcula japonica strain TR-1 (HjDHFR P1) (Miyashita et al. 2015). H. japonica TR-1 was isolated from a saltern at the Noto peninsula in Ishikawa, Japan, and requires 41–650 mM Mg2+ and high concentrations (1.7–4.3 M) of NaCl for growth (Hamamoto et al. 1988; Nishiyama et al. 1992; Horikoshi et al. 1993; Nakamura et al. 2010). Three DHFR genes, which express HjDHFR C1, C2, and P1, are coded in the genome of this archaeon. We showed that the addition of NaCl up to 500 mM induced the structural formation of HjDHFR P1 at pH 8 using circular dichroism (CD) and fluorescence spectra, but the same structure was formed without NaCl at pH 6 or by the addition of NADPH. Nevertheless, NaCl enhanced the activity of HjDHFR P1 at pH 6, showing maximum activity at approximately 500 mM. Conversely, the further addition of NaCl reduced its activity, although its structural stability increased monotonously up to 1000 mM. We proposed the binding of anions as the mechanism underlying the activation of HjDHFR P1 by salt; however, the detailed mechanisms underlying its activation and inactivation are unclear. The purpose of this work was to elucidate how salt ions affect the enzymatic function of HjDHFR P1 during its activation and inactivation processes. In this paper, we evaluate the effects of salt on the elementary steps in the catalytic cycle of HjDHFR P1 by monitoring the pH- and salt concentration dependences of its enzyme activity, deuterium isotope effects, and rapid-phase ligand-binding kinetics using stopped-flow fluorescence quenching. On the basis of the results of these experiments, we discuss the activation and inactivation mechanisms of HjDHFR P1 by salt and compare the effects of salt with other halophilic DHFRs, namely, HvDHFR 1 and 2, and non-halophilic EcDHFR.
Materials and methods
Protein expression and purification
The expression and purification methods for HjDHFR P1 were described previously (Miyashita et al. 2015). The concentration of purified HjDHFR P1 was determined spectrophotometrically using a molar extinction coefficient of 20,910 M−1 cm−1 at 280 nm.
pH dependence of enzyme activity
The pH dependence of the enzyme activity of HjDHFR P1 was evaluated spectrophotometrically using a V-560 spectrophotometer (JASCO, Inc., Tokyo, Japan). The initial velocity of the enzymatic reaction was determined by monitoring the change of absorbance using a molar extinction coefficient of 11,800 M−1 cm−1 at 340 nm (Stone and Morrison 1984). The buffer used was 50 mM 2-(N-morpholino)ethanesulfonic acid, 25 mM Tris, 25 mM ethanolamine, 0.1 mM EDTA, and 0.1 mM dithiothreitol (MTE buffer) containing 0, 200, or 1000 mM NaCl, whose pH was adjusted by acetic acid or tetramethylammonium hydroxide (TMAOH). The reaction solution without DHF was pre-incubated for 10 min at 25 °C, and the reaction was initiated by the addition of the DHF solution, which was also pre-incubated at 25 °C. The temperature of the reaction mixture was maintained at 25 °C with a circulating thermobath (NESLAB RTE-5; Thermo Fisher Scientific, Waltham, MA). The concentrations of DHF (Sigma–Aldrich, St. Louis, MO) and NADPH (Oriental Yeast, Tokyo, Japan) were determined spectrophotometrically using molar extinction coefficients of 28,000 M−1 cm−1 at 282 nm and 6200 M−1 cm−1 at 339 nm, respectively. The concentrations of the enzyme, NADPH, and DHF in the reaction mixture were 0.08, 50, and 50 μM, respectively. Observed initial velocity (V) was plotted against pH and fitted to the following equation (Stone and Morrison 1984):
where V i is the pH-independent velocity, K a and K b are acid dissociation constants, and [H+] is proton concentration.
Effects of salt concentration on enzyme activity
The effects of NaCl, tetramethylammonium chloride (TMACl), and sodium acetate (CH3COONa) concentrations on the enzyme activity of HjDHFR P1 were measured at 25 °C and pH 8.0. The effect of NaCl concentration was also measured at pH 6.0 and 10.0. The buffer used and concentrations of the enzyme, NADPH, and DHF were the same as for the pH-dependence measurements. The initiating method of the reaction was also the same as for the pH-dependence measurements.
Deuterium isotope effects on steady-state kinetics
4(R)-2H reduced NADPH (NADPD) was synthesized from NADP+ (Oriental Yeast) and 2-propanol-d8 (Sigma–Aldrich) by the catalytic reaction of alcohol dehydrogenase from Thermoanaerobium brockii (Sigma–Aldrich) as reported previously (Chen et al. 1987). Synthesized NADPD was purified using AG MP-1 anion exchange resin (BIO-RAD, Hercules, CA) by the method of Viola et al. (1979), and stored at −20 °C. Just before use, NADPD was thawed and desalted by a HiTrap desalting column (GE Healthcare Japan, Tokyo, Japan), and its purity and concentration were confirmed by absorptions at 260 and 340 nm. NADPH used in this experiment was also prepared by the same method to normalize the effects of contaminating salt ions. The deuterium isotope effect in the steady-state catalytic reaction, D V, was evaluated as follows (David et al. 1992):
where V NADPH and V NADPD are the initial velocities of the enzymatic reaction using NADPH and NADPD as a cofactor, respectively. The buffer used and concentrations of the enzyme, cofactors, and DHF were the same as for the pH-dependence measurements.
Steady-state enzyme kinetics
Steady-state enzyme kinetics were measured at 25 °C and pH 6.0 or 8.0. The buffer used was MTE buffer. When the kinetics parameters for DHF were measured, the concentration of DHF was varied from 0 to 100 μM, employing 50 μM NADPH. When those for NADPH were measured, the concentration of NADPH was varied from 0 to 150 μM, employing 50 μM DHF. Enzyme concentrations were 10–200 nM, depending on the pH and NaCl concentration, and determined by a methotrexate (MTX) titration method (Williams et al. 1979). Reaction solutions without DHF or NADPH were pre-incubated for 10 min before initiating the reaction by the addition of the pre-incubated substrate solution.
Effects of NaCl concentration during pre-incubation
The effects of NaCl concentration during pre-incubation were measured at 25 °C and pH 6.0. The buffer used and final concentrations of the enzyme, NADPH, and DHF were the same as for the pH-dependence measurements. Enzyme and NADPH or DHF were pre-incubated for 10 min under 0 to 2000 mM NaCl, and 100 μL of the solution were added to 900 μL of the pre-incubated substrate solution containing an appropriate concentration of NaCl for a final concentration of 500 mM. Reaction rate was calculated from absorption change from 60 to 90 s, since that from 0 to 60 s was nonlinear for several conditions. Then, it was plotted against NaCl concentration during pre-incubation, and fitted to the following equation:
where V max is the maximum velocity, K d is the dissociation constant between enzyme and salt, and V 0 is the NaCl concentration-independent velocity, which is needed to correct the effects of 500 mM NaCl included in the reaction mixture.
Rapid-phase ligand-binding kinetics
The rapid-phase ligand-binding kinetics of DHF and NADPH to HjDHFR P1 at various NaCl concentrations were measured by a fluorescence quenching method using a model SX20 stopped-flow system (Applied Photophysics, Surrey, UK). Three intrinsic tryptophan side chains were excited at 280 nm, and fluorescence at approximately 350 nm was detected using a V-350 bandpass filter (Hokushin Optical Works Ltd., Tokyo, Japan). The buffer used was MTE buffer (pH 6.0), and temperature was maintained at 24.5 °C. The final concentrations of the enzyme, DHF, and NADPH in the reaction mixture were 4.8, 50, and 25 μM, respectively. The resulting fluorescence intensity as a function of time, F(t), was fitted to a single exponential with linear decay:
where A, k app, k lin, and F ∞ are the amplitude of fluorescence quenching, apparent rate constants for the exponential and linear phases, and finally attaining fluorescence intensity, respectively. Since the obtained k lin values were negligibly small for the DHF-binding measurements, fitting was repeated with this parameter fixed to zero.
To determine ligand-association and -dissociation rate constants, the concentration of HjDHFR P1 was reduced to 1 μM and those of the ligands were varied from 10 to 50 μM. Then, the fluorescence decay data were fitted to Eq. 4, and the obtained k app was fitted to the following equation (Grubbs et al. 2011):
where k on and k off are the association and dissociation rate constants, respectively, and [ligand] indicates ligand concentration. The rate constants for THF were measured at pH 8.0, and the k lin parameter was fixed to zero once again for the THF-binding measurements.
Results
pH and salt dependences of enzyme activity
Figure 1a shows the pH dependence of the enzyme activity of HjDHFR P1 in the absence or presence of 200 or 1000 mM NaCl as a logarithmic scale. The apparent pK a and pK b values obtained from fittings using Eq. 1 are listed in Table 1. The activity of HjDHFR P1 without NaCl was obviously pH dependent; it was almost constant at the neutral pH region (5.2–8.0) and clearly decreased at the higher and lower pH regions with pK a and pK b values of 5.0 ± 0.2 and 8.4 ± 0.2, respectively. It is known that EcDHFR shows similar pH-dependent enzyme activity at neutral to basic pH region, and the rate-determining step of the enzymatic reaction is changed from the THF-releasing step at the neutral pH region to the hydride-transfer step at a pH greater than 8.4 (Fierke et al. 1987). Therefore, similarly to EcDHFR, the rate-determining step of HjDHFR P1 above pH 8.4 was presumed to be the hydride-transfer step. However, the rate-determining step from the acidic to neutral pH region (pH 5.0–8.0) was unclear, because each of the two binding and two releasing steps could be a candidate, although the former could be eliminated if the DHF and NADPH concentrations in this experimental condition (both 50 μM) are excessive. The addition of NaCl clearly increased the enzymatic activity of HjDHFR P1 from the acidic to neutral pH region, but only had small effects above pH 8.5. Thus, NaCl accelerated the reaction rate of the rate-determining step at the acidic to neutral pH region, but did not enhance that at the basic pH region. The decrease of pK b values from 8.4 to 7.4 by the addition of NaCl also indicated that the hydride-transfer step became the rate-determining step at pH 7.4–8.4 by accelerating the rate-determining step at this pH region. Conversely, decreased enzyme activity at pH 5 in the absence and presence of NaCl, which is not observed for EcDHFR, might suggest the contribution of another dissociable group to the enzymatic reaction of HjDHFR P1. However, aggregation of the enzyme could contribute because calculated pI of this enzyme is 4.8, and the reason for this observation was unclear because of poor data points at the acidic pH region.

a pH dependence of the enzymatic activity of HjDHFR P1 at 25 °C under 0 (black), 200 (red), and 1000 mM (green) NaCl. The buffer used was MTE buffer whose pH was adjusted by acetic acid or TMAOH. The lines indicate the nonlinear least squares fits to Eq. 1 (see “Materials and methods”). b NaCl concentration dependence of the enzymatic activity of HjDHFR P1 at 25 °C and pH 6.0 (black), 8.0 (red), and 10.0 (green). Inset of b NaCl (black), TMACl (red), and CH3COONa (green) concentration dependences of the enzymatic activity of HjDHFR P1 at 25 °C and pH 8.0
Figure 1b shows the NaCl concentration dependence of the enzyme activity of HjDHFR P1 at pH 6.0, 8.0, and 10.0. At pH 6.0, enzyme activity was enhanced approximately eightfold by the addition of 500 mM NaCl, and conversely decreased by the further addition of NaCl. At pH 8.0, only a fourfold enhancement was observed at 250 mM NaCl, and the activity decreased gradually at higher concentrations of NaCl. Finally, the enzyme activity of HjDHFR P1 became independent of NaCl concentration at pH 10.0 (expanded figure was provided as Fig. S1). This result clearly indicated that the reaction rate of the rate-determining step at the neutral pH region depended on NaCl concentration, and that at pH 10 was independent of NaCl concentration. In addition, we measured the NaCl, TMACl, and CH3COONa concentration dependences of the enzyme activity of HjDHFR P1 (inset of Fig. 1b). The difference in the activation effects between organic and inorganic cations and anions clearly indicated that chloride anions activated HjDHFR P1.
Deuterium isotope effects on steady-state kinetics
To clarify the relationship between the solvent environment (pH and NaCl concentration) and hydride-transfer rate in the catalytic cycle of HjDHFR P1, we measured deuterium isotope effects on steady-state enzyme activity. The isotope effects, D V, were evaluated by calculating the ratio of the initial velocities between using NADPH and NADPD as a cofactor, and are listed in Table 2. David et al. (1992) reported that D V values in the steady-state turnover of EcDHFR could range from 1 to 3, and if the rate-determining step is hydride transfer, the D V value is close to 3. Conversely, if the rate-determining step is 3 times slower than the hydride-transfer rate, the D V value is close to 1. As shown in Table 2, the D V values of HjDHFR P1 at pH 10.0 were 2.7–3.3, indicating that hydride transfer was fully rate determining independent to NaCl concentration. This value was decreased to 1.0–1.2 at pH 6.0, indicating that hydride transfer was sufficiently faster than the rate-determining step at this pH. Conversely, the D V value at pH 8.0 increased from 1.5 to 2.8 as NaCl concentration increased from 0 to 1000 mM, indicating that hydride transfer changed from partially rate-determining to fully rate-determining as the NaCl concentration increased. These data clearly showed that the activation mechanism of HjDHFR P1 by salt was unrelated to hydride transfer from NADPH to DHF.
Steady-state enzyme kinetics
To confirm that the DHF- and NADPH-binding steps could be eliminated as candidates for the rate-determining step at the neutral pH region, we determined the steady-state kinetics parameters of the enzymatic reaction of HjDHFR P1 at 25 °C and pH 6.0 or 8.0 under several concentrations of NaCl. The obtained parameters are listed in Table 3. At pH 6.0, the k cat values for DHF and NADPH increased drastically from 3 to 20 s−1 as the NaCl concentration increased from 0 to 500 mM, similar to the activation profile of HjDHFR P1 at this pH (Fig. 1b). However, the K m values for DHF and NADPH increased fourfold and only slightly, respectively, by the addition of 500 mM NaCl. It is noteworthy that the maximum K m values (8.1 ± 0.7 μM for both DHF and NADPH) were less than 10 μM. Thus, the DHF and NADPH concentrations used for the measurements of pH and salt concentration dependences (both 50 μM) could be considered to be in excess, and the binding steps of both substrates were not the rate-determining step at pH 6.0. Conversely, at pH 8.0, the k cat values for DHF and NADPH only increased slightly from 1.5 to 3 s−1 as the NaCl concentration increased from 0 to 200 mM and decreased gradually at higher NaCl concentrations, which also matched the activation profile at this pH (Fig. 1b). In addition, the K m values were almost independent of NaCl concentration considering experimental error, and the maximum values (8.5 ± 1.4 μM and 6.4 ± 1.1 μM for DHF and NADPH, respectively) were still less than 10 μM. Thus, the concentration of 50 μM could also be assumed to be in excess, and the binding steps were also not the rate-determining step at pH 8.0.
Effects of NaCl concentration during pre-incubation
To check the maximum activation effect of chloride anions, we measured the enzyme activity of HjDHFR P1 at the pH 6.0 and 500 mM NaCl condition employing pre-incubation with various concentrations of NaCl. Figure 2a shows the time courses of the absorption changes initiated by the addition of the pre-incubated solution containing enzyme, NADPH, and various concentrations of NaCl to the DHF solution containing an appropriate concentration of NaCl to a final concentration of 500 mM. As shown in Fig. 2a, the initial slope of the absorption change increased and the reaction ended faster as the NaCl concentration increased during pre-incubation. The reaction rates of the enzymatic reaction were calculated from the slopes of 60–90 s, and plotted against the NaCl concentration during pre-incubation (inset of Fig. 2a). The reaction rate increased gradually as the NaCl concentration during pre-incubation increased from 0 to 1000 mM, and became almost constant over 1250 mM. It is noteworthy that the reaction rate increased continuously at more than 500 mM NaCl, in which the maximum activity is observed at this pH (Fig. 1b). This result clearly indicated that the activation effect of chloride anions continued over 500 mM, but higher concentrations of NaCl in the reaction mixture reduced the steady-state turnover rate of HjDHFR P1. To determine the dissociation constant, K d, between the enzyme and chloride anion, the data were fitted to Eq. 3, which were derived from one-to-one binding of the enzyme and anion. The calculated K d value was 1430 ± 740 mM. Although this value seemed inaccurate due to activity measurements in the presence of 500 mM NaCl, its successful fitting indicated that binding of a chloride anion to HjDHFR P1 enhanced enzymatic activity.

Time course of the absorption change at 340 nm due to the enzymatic reaction of HjDHFR P1 at 25 °C and pH 6.0 as a function of NaCl concentration during pre-incubation. The enzymatic reaction was initiated by the addition of an enzyme-DHF-NaCl mixture to an NADPH solution containing NaCl to a final concentration of 500 mM (a) or an enzyme-NADPH-salt mixture to a DHF solution containing NaCl to a final concentration of 500 mM (b). The buffer used was MTE buffer. NaCl concentrations during pre-incubation are indicated by the following colors: 0 (black), 250 (red), 500 (green), 750 (blue), 1000 (cyan), 1250 (magenta), 1500 (yellow), 1750 (orange), and 2000 mM (dark green). Insets Plots of initial velocities as a function of NaCl concentration during pre-incubation. The solid lines are least squares fits to Eq. 3 derived by the simple one-to-one binding model (see “Materials and methods”)
When the reaction was initiated by the addition of the enzyme-DHF-salt mixture to the NADPH solution, similar results were also observed, and the calculated K d value was 1980 ± 850 mM (Fig. 2b). However, it was noteworthy that when the enzyme-DHF solution without NaCl was mixed with the NADPH solution containing NaCl, the initial slope of the absorbance change at 0 s was very low. Then, the slope increased gradually as the reaction progressed up to 60 s and became constant. This observation indicated a slight acceleration of the enzyme reaction during the reaction period. Such an activation effect was not observed when the NaCl concentration during pre-incubation was more than 500 mM or the reaction was initiated by mixing the enzyme-NADPH and DHF solutions. Therefore, it seemed that DHF could not bind to HjDHFR P1 without NADPH or chloride anions. To confirm this point, we measured rapid-phase ligand-binding kinetics.
Rapid-phase ligand-binding kinetics
To evaluate the effects of salt on the substrate-binding reactions of HjDHFR P1, we monitored the rapid-phase (shorter than 1 s) fluorescence quenching of intrinsic tryptophan side chains by the binding of DHF or NADPH at pH 6.0 and 24.5 °C under various NaCl concentrations. Figure 3 shows typical results of the time courses of the fluorescence quenching by binding of DHF (Fig. 3a) or NADPH (Fig. 3b) to the enzyme. As shown in Fig. 3a, the change in fluorescence due to binding of DHF was very small in the absence of NaCl, and it became large as NaCl concentration increased, although the concentrations of the enzyme and DHF were not changed. This result suggested that salt enhanced the binding of DHF to HjDHFR P1. The observed fluorescence decay data were fitted to Eq. 4, and the obtained apparent rate constant, k app, and amplitude, A, were plotted against NaCl concentration (Table S1, inset of Fig. 3a). The A value obviously increased as NaCl concentration increased, similar to the activation effect of chloride anions (inset of Fig. 2a). However, k app was almost independent of NaCl concentration in the range of 16 ± 7 s−1. These results indicated that DHF could only bind to chloride anion-bound HjDHFR P1 molecules, but the binding rate of DHF was independent of NaCl concentration.

Quenching of intrinsic tryptophan fluorescence by rapid binding of DHF (a) or NADPH (b) to HjDHFR P1 at 24.5 °C and pH 6.0 under various NaCl concentrations. The buffer used was MTE buffer and the concentrations of enzyme, DHF, and NADPH were 4.8, 50, and 25 μM, respectively. Black, red, green, blue, and cyan indicate 0, 250, 500, 1000, and 2000 mM NaCl, respectively. Insets NaCl concentration dependence of the apparent rate constant (black) and amplitude parameter (red) of the exponential phase obtained by fitting to Eq. 4 (see “Materials and methods”)
Conversely, fluorescence quenching by the binding of NADPH was very rapid in the absence of NaCl, and became slower as NaCl concentration increased (Fig. 3b). The k app value was decreased by 20-fold from 365 ± 68 to 16 ± 0 s−1 as NaCl concentration increased from 250 to 2000 mM (inset of Fig. 3b). The A value only increased by fourfold from 0.19 ± 0.03 to 0.72 ± 0.03 as NaCl concentration increased from 250 to 1000 mM, and slightly decreased under higher concentrations of NaCl. These results suggested that NADPH bound rapidly to chloride anion-unbound HjDHFR P1 molecules, and more slowly to the chloride anion-bound enzyme.
Association and dissociation rate constants of ligands
To obtain information for the effects of salt on the binding and releasing rates of ligands, the ligand concentration dependence of the k app value was measured under various NaCl concentrations (Tables S2–S4). As shown in Fig. 4a, the k app value increased as DHF concentration increased. Similar results were also obtained for NADPH (Fig. 4b). However, fluorescence quenching by the binding of NADP+ and THF is difficult to measure at pH 6.0; therefore, pH was raised to 8.0. As a result, THF binding became measurable (Fig. 4c), but the binding of NADP+ to HjDHFR P1 could not be measured even at pH 8.0. From the slope and intercept of these plots, association and dissociation rate constants, k on and k off, respectively, were determined, plotted against NaCl concentration in the insets of Fig. 4, and are listed in Table 4. As shown in the insets of Fig. 4a, c, the k on values for DHF and THF seemed almost independent of NaCl concentration; however, the k off values for THF decreased slightly as NaCl concentration increased, consistent with the decrease of the k cat value at pH 8.0 (Table 3). This result suggested that the THF-releasing step is the rate-determining step of HjDHFR P1 at the neutral pH region, similar to EcDHFR.

NaCl concentration dependence of association (black) and dissociation (red) rate constants for DHF (a), NADPH (b), and THF (c) to HjDHFR P1 at 24.5 °C. The buffer used was MTE buffer whose pH was adjusted to 6.0 (a, b) or 8.0 (c) by acetic acid or TMAOH. Insets Ligand concentration dependence of the apparent rate constants for the binding at various NaCl concentrations. NaCl concentrations are indicated by the following colors: 250 (black), 500 (red), 1000 (green), 1500 (blue), and 2000 mM (cyan). The lines indicate least squares linear fits
Conversely, those for NADPH were significantly, approximately 10-fold, decreased as NaCl concentration increased from 500 to 2,000 mM (inset of Fig. 4b). Nevertheless, the k on value of 0.42 ± 0.01 μM−1 s−1 at 2000 mM NaCl generated a binding rate of 21 s−1 for 50 μM NADPH. This rate is substantially faster than the rate-determining step at pH 6.0 and 2000 mM NaCl (approximately 3 s−1); therefore, the NADPH-binding step is not the rate-determining step of HjDHFR P1 at the neutral pH region. This speculation is also confirmed by the sufficiently small K m value for NADPH (5.7 ± 0.5 μM), as mentioned above, although this value was measured at pH 8.0 and 1000 mM NaCl (Table 3).
Discussion
The enzymatic activity of moderately halophilic enzymes shows activation by salt and following inactivation by the same salt. Such an observation is confusing because the same salt has opposite effects on the same enzyme depending on its concentration. However, the steady-state turnover of these enzyme contains various enzyme-ligand complexes and multiple steps between them. Therefore, salt can affect the equilibria and reaction rates for these complexes in different ways, and the population of the complexes and the rate-determining step of enzyme turnover can be changed depending on the salt concentration, resulting in the inversion of the apparent effect of salt on activity. The purpose of this study was to clarify such activation and inactivation mechanisms on the enzyme activity of HjDHFR P1 by salt, and discuss them in comparison with those of other halophilic DHFRs: HvDHFR 1 and 2, and non-halophilic EcDHFR.
Activation mechanism of HjDHFR P1 by salt
From a comparison of the effects of inorganic and organic cations and anions, it is obvious that chloride anions enhance the enzymatic activity of HjDHFR P1 (inset of Fig. 1b). Furthermore, the hyperbolic activation effects against salt concentration shown in the insets of Fig. 2 indicated that anion binding was crucial. Although anion can bind to both the enzyme and substrates, the effective binding is occurred on the enzyme or enzyme-ligand complexes since the activation effect was observed in the saturated substrates condition. However, there are two possibilities for such an activation mechanism induced by anion binding: (1) acceleration of the rate-determining step in the catalytic cycle, and (2) population change of the inactive (or low-activity) and active (or high-activity) conformers caused by the equilibrium shift between them.
The steady-state enzymatic turnover of DHFR includes at least five steps: two binding steps of NADPH and DHF, hydride transfer from NADPH to DHF, and two releasing steps of NADP+ and THF. From the sufficiently small K m values observed in the steady-state kinetics experiments, the two binding steps can be eliminated as candidates for the rate-determining step (Table 3). The constant enzyme activity (Fig. 1b) and full isotope effects (Table 2) at pH 10.0 indicate that the hydride-transfer rate is independent of salt concentration, and the activation mechanism affects the rate-determining step at the neutral pH region. In addition, from the rapid-phase ligand-binding experiments, the binding rates of DHF and NADPH to HjDHFR P1 and the releasing rate of THF from the enzyme were not accelerated by salt (Table 4). Although we could not measure the NADP+-releasing rate, the difficulty of this measurement suggested a rapid reaction rate for this process and low probability that this step was the rate-determining step. Therefore, mechanism (1) seems improbable.
Conversely, when the enzymatic reaction was initiated by mixing the enzyme-DHF solution without salt to the NADPH solution containing salt, the initial activity of HjDHFR P1 was low and increased gradually as the reaction progressed (Fig. 2b). In addition, the amplitude of fluorescence quenching by the rapid binding of DHF clearly increased with increasing salt concentration (inset of Fig. 3a). These results indicate that DHF cannot bind to HjDHFR P1 before the anion binds to the enzyme. Therefore, mechanism (2), population change of the anion-bound and anion-unbound enzyme conformers, which are the binding-competent and -incompetent forms for DHF, respectively, is a reasonable explanation for the activation mechanism of HjDHFR P1 by salt, although we previously reported that the secondary structure of HjDHFR P1 is already formed in the absence of salt at pH 6.0 (Miyashita et al. 2015).
Such ligand-binding-competent and -incompetent conformers are also observed for EcDHFR as E1 and E2, respectively (Cayley et al. 1981). In the case of EcDHFR, the exponential phase shown in the rapid-phase fluorescence quenching reflects the binding of NADPH, DHF, or folate to the E1 conformer, and the subsequent linear phase reflects interconversion from E2 to E1. Since the binding of NADPH to HjDHFR P1 showed similar exponential and linear phases (Table S1), it is possible that the latter phase reflect the interconversion of the binding-competent and -incompetent conformers. However, such interconversion could be negligible for the salt concentration dependence of the enzyme activity shown in Fig. 1b, because we pre-incubated the enzyme with NADPH for 10 min, and the pre-incubation from 5 to 20 min showed almost the same results (Miyashita et al. 2015). Conversely, binding of DHF to HjDHFR P1 showed only single exponential phase suggesting that interconversion between DHF binding-competent and -incompetent conformers seems hardly occurred without salt, although additional measurements in longer time scale are needed.
Inactivation mechanism of HjDHFR P1 by salt
As discussed above, the activation mechanism of HjDHFR P1 by salt could be explained by the population change of the active and inactive forms of the enzyme. Thus, the rate-determining step of enzyme turnover need not change according to salt concentration. Therefore, it is presumable that the rate-determining step at the neutral pH region is the THF-releasing step, as for EcDHFR, and the rate of this step was decelerated by salt. The consistency between the k cat and k off values for THF confirms this presumption (Tables 3, 4). We previously suggested that the inactivation profile of HjDHFR P1 is exponential and not explained by the binding of salt (Miyashita et al. 2015). The deceleration of the THF-releasing step is consistent with this observation.
Salt effects on the elementary steps of the enzymatic reaction of HjDHFR P1
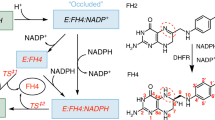
From the experimental results of this report, we summarized the salt effects on the elementary steps of the enzymatic reaction of HjDHFR P1 as shown in Fig. 5. Anion-unbound enzyme and anion-bound enzyme are in equilibrium in solution. The NADPH-binding rate for the former conformer is more rapid than for the latter one. DHF can bind only to the latter conformer, but the reaction rate is salt concentration independent. During steady-state turnover, hydride transfer from NADPH to DHF, which is the rate-determining step at the basic pH region, is independent of salt concentration. In addition, the THF-releasing step, which is the rate-determining step at the neutral pH region, is decelerated by salt. Although anion-unbound enzyme can create another catalytic cycle, as we assumed in a previous report, the catalytic efficiency of this conformer is very low compared with the anion-bound conformer and it can be ignored.

Schematic drawing of the effects of salt on the elementary steps of the enzymatic reaction of HjDHFR P1 at the neutral pH region. Objects shown in gray and pink indicate the anion-unbound and anion-bound conformers, respectively. Triangle symbols represent DHF (orange) and THF (red), respectively. Star symbols represent NADPH (green) and NADP+ (light blue), respectively. The red and blue arrows indicate salt concentration-dependent and -independent processes, respectively. Thick arrows indicate the major reaction pathway
Comparison with other DHFRs
It is noteworthy that the activation mechanism of HjDHFR P1 is consistent with previous results for other halophilic DHFRs (i.e., HvDHFR 1 and 2). Salt induces the structural formation of both HvDHFRs resulting in an enhancement of their enzymatic activity (Wright et al. 2002). The equilibrium and kinetic stability studies of HvDHFR 1 show that structural formation indicates a population increase of the number of folded molecules caused by destabilization of the unfolded state (Gloss et al. 2008). However, the stabilizing effect of salt is not specific for halophilic enzymes; the same effect is also observed for EcDHFR (Wright et al. 2002). In addition, the structural formation of HvDHFR 1 is almost complete at 1 M KCl, as determined by monitoring CD and fluorescence spectra, although it is activated monotonously up to 3.5 M KCl (Wright et al. 2002). Thus, HvDHFR 1 should have another activation mechanism. Blecher et al. (1993) reported that the K m value for DHF is decreased by tenfold from 0.9 to 0.08 mM as KCl concentration increases from 0.5 to 3.0 M, indicating the enhancement of the affinity between DHF and HvDHFR 1. Such an observation can be explained by the existence of binding-competent and -incompetent conformers for DHF. Conversely, HvDHFR 2 shows similar salt concentration dependence of enzyme activity to HjDHFR P1, with a maximum at 500 mM and gradual decrease by the further addition of KCl. Although the detailed activation and inactivation mechanisms are not clear, the same mechanisms can be presumed.
Since the rate-determining step of EcDHFR at the neutral pH region is the THF-releasing step, EcDHFR should also show a similar salt-inactivation profile to HjDHFR P1. However, according to previous reports, the enzymatic activity of EcDHFR is markedly decreased as NaCl concentration increases (Baccanari et al. 1975; Ohmae et al. 2013). This is caused by the binding of an inorganic cation near the substrate-binding cleft, which was confirmed by NMR experiments. Although inorganic cations strongly inhibit the enzyme activity of EcDHFR, TMACl, which has an organic cation, induces a gradual decrease of its activity, an approximately 30% reduction from 0 to 500 mM at pH 8.0, consistent with the inactivation effect of NaCl on HjDHFR P1 (Ohmae et al. 2013). Therefore, deceleration of the THF-releasing rate by salt may be a common feature of both DHFRs, and HjDHFR P1 may have maximum activity in the absence of salt if the population change of the active conformer has not occurred.
Conclusions
HjDHFR P1 shows moderately halophilic characteristics for enzymatic activity at pH 6.0, although there are no significant effects of NaCl on its secondary structure, as monitored by CD spectra. pH and salt concentration dependencies showed that the enzyme was activated at the acidic to neutral pH region, but not activated at the basic pH region, in which the rate-determining step was the hydride-transfer step. Conversely, rapid-phase ligand-binding experiments using stopped-flow fluorescence quenching showed that the amplitude of the rapid binding of DHF to HjDHFR P1 increased with increasing NaCl concentration at pH 6.0, although the reaction rate was almost constant. In addition, the THF-releasing rate decreased with increasing NaCl concentration, consistent with the decrease of the k cat value. These results suggested that the activation mechanism of HjDHFR P1 by salt is population change of anion-unbound and anion-bound conformers, which are binding-incompetent and -competent forms for DHF, respectively. On the other hand, the salt-inactivation mechanism is via deceleration of the THF-releasing rate, which is the rate-determining step at the neutral pH region. This activation mechanism may also be possible for other halophilic DHFRs—HvDHFR 1 and 2—and the inactivation mechanism may be a common feature of non-halophilic EcDHFR.
Abbreviations
- CD:
-
Circular dichroism
- DHF:
-
Dihydrofolate
- DHFR:
-
Dihydrofolate reductase
- EcDHFR:
-
DHFR from Escherichia coli
- HjDHFR:
-
DHFR from Haloarcula japonica
- HvDHFR:
-
DHFR from Haloferax volcanii
- MTE:
-
50 mM 2-(N-morpholino)ethanesulfonic acid, 25 mM Tris, and 25 mM ethanolamine containing 0.1 mM dithiothreitol and 0.1 mM EDTA (buffer)
- MTX:
-
Methotrexate
- NADPD:
-
4(R)-2H reduced nicotinamide adenine dinucleotide phosphate
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NMR:
-
Nuclear magnetic resonance
- THF:
-
Tetrahydrofolate
- TMACl:
-
Tetramethylammonium chloride
- TMAOH:
-
Tetramethylammonium hydroxide
References
Baccanari D, Phillips A, Smith S, Sinski D, Burchall J (1975) Purification and properties of Escherichia coli dihydrofolate reductase. Biochemistry 14:5267–5273
Behiry EM, Evans RM, Guo J, Loveridge EJ, Allemann RK (2014) Loop interactions during catalysis by dihydrofolate reductase from Moritella profunda. Biochemistry 53:4769–4774
Binbuga B, Boroujerdi AFB, Young JK (2007) Structure in an extreme environment: NMR at high salt. Protein Sci 16:1783–1787
Blecher O, Goldman S, Mevarech M (1993) High expression in Escherichia coli of the gene coding for dihydrofolate reductase of the extremely halophilic archae bacterium Haloferax volcanii. Eur J Biochem 216:199–203
Boroujerdi AFB, Young JK (2009) NMR-derived folate-bound structure of dihydrofolate reductase 1 from the halophile Haloferax volcanii. Biopolymers 91:140–144
Cayley PJ, Dunn SM, King RW (1981) Kinetics of substrate, coenzyme, and inhibitor binding to Escherichia coli dihydrofolate reductase. Biochemistry 20:874–879
Chen J, Taira K, Tu CD, Benkovic SJ (1987) Probing the functional role of phenylalanine-31 of Escherichia coli dihydrofolate reductase by site-directed mutagenesis. Biochemistry 26:4093–4100
David CL, Howell EE, Farnum MF, Villafranca JE, Oatley SJ, Kraut J (1992) Structure and function of alternative proton-relay mutants of dihydrofolate reductase. Biochemistry 31:9813–9822
Fierke CA, Johnson KA, Benkovic SJ (1987) Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry 26:4085–4092
Garvey EP, Matthews CR (1989) Effects of multiple replacements at a single position on the folding and stability of duhydrofolate reductase from Escherichia coli. Biochemistry 28:2083–2093
Gloss LM, Topping TB, Binder AK, Lohman JR (2008) Kinetic folding of Haloferax volcanii and Escherichia coli dihydrofolate reductases: Haloadaptation by unfolded state destabilization at high ionic strength. J Mol Biol 376:1451–1462
Grubbs J, Rahmanian S, DeLuca A, Padmashali C, Jackson M, Duff MR Jr, Howell E (2011) Thermodynamics and solvent effects on substrate and cofactor binding in Escherichia coli chromosomal dihydrofolate reductase. Biochemistry 50:3673–3685
Guo J, Luk LYP, Loveridge EJ, Allemann RK (2014) Thermal adaptation of dihydrofolate reductase from the moderate thermophile Geobacillus stearothermophilus. Biochemistry 53:2855–2863
Hamamoto T, Takashina T, Grant WD, Horikoshi K (1988) Asymmetric cell division of a triangular halophilic archaebacterium. FEMS Microbiol Lett 56:221–224
Horikoshi K, Aono R, Nakamura S (1993) The triangular halophilic archaebacterum Haloarcula japonica strain TR-1. Experientia 49:497–502
Jennings PA, Finn BE, Jones BE, Matthews CR (1993) A reexamination of the folding mechanism of dihydrofolate reductase from Escherichia coli: verification and refinement of a four-channel model. Biochemistry 32:3783–3789
Kuwajima K, Garvey EP, Finn BE, Matthews CR, Sugai S (1991) Transient intermediates in the folding of dihydrofolate reductase as detected by far-ultraviolet circular dichroism spectroscopy. Biochemistry 30:7693–7703
Luk LYP, Loveridge EJ, Allemann RK (2014) Different dynamical effects in mesophilic and hyperthermophilic dihydrofolate reductases. J Am Chem Soc 136:6862–6865
Miyashita Y, Ohmae E, Nakasone K, Katayanagi K (2015) Effects of salt on the structure, stability, and function of a halophilic dihydrofolate reductase from a hyper halophilic archaeon, Haloarcula japonica strain TR-1. Extremophiles 19:479–493
Nakamura S, Nakasone K, Takashina T (2010) Genetics and genomics of triangular disc-shaped halophilic archaeon Haloarcula japonica strain TR-1. In: Horikoshi K, Antranikian G, Bull A, Robb F, Stetter K (eds) Extremophiles Handbook. Springer, Tokyo, pp 363–381
Nishiyama Y, Takashina T, Grant WD, Horikoshi K (1992) Ultrastructure of the cell wall of the triangular halophilic archaebacterum Haloarcula japonica strain TR-1. FEMS Microbiol Lett 99:43–48
Ohmae E, Murakami C, Tate S, Gekko K, Hata K, Akasaka K, Kato C (2012) Pressure dependence of activity and stability of dihydrofolate reductases of the deep-sea bacterium Moritella profunda and Escherichia coli. Biochim Biophys Acta 1824:511–519
Ohmae E, Miyashita Y, Tate S, Gekko K, Kitazawa S, Kitahara R, Kuwajima K (2013) Solvent environments significantly affect the enzymatic function of Escherichia coli dihydrofolate reductase: comparison of wild-type protein and active-site mutant D27E. Biochim Biophys Acta 1834:2782–2794
Ortenberg R, Rozenblatt-Rosen O, Mevarech M (2000) The extremely halophilic archaeon Haloferax volcanii has two very different dihydrofolate reductases. Mol Microbiol 35:1493–1505
Osborne MJ, Venkitakrishnan RP, Dyson HJ, Wright PE (2003) Diagnostic chemical shift markers for loop conformation and substrate and cofactor binding in dihydrofolate reductase complexes. Protein Sci 12:2230–2238
Perry KM, Onuffer JJ, Touchette NA, Herndon CS, Gitteleman MS, Matthews CR, Chen JT, Mayer RJ, Taira K, Benkovic SJ, Howell EE, Kraut J (1987) Effects of single amino acid replacements on the folding and stability of dihydrofolate reductase from Escherichia coli. Biochemistry 26:2674–2682
Pieper U, Kapadia G, Mevarech M, Herzberg O (1998) Structural features of halophilicity derived from the crystal structure of dihydrofolate reductase from the Dead Sea halophilic archaeon, Haloferax volcanii. Structure 6:75–88
Roesser M, Müller V (2001) Osmoadaptation in bacteria and archaea: common principles and differences. Environ Microbiol 3:743–754
Sawaya MR, Kraut J (1997) Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry 36:586–603
Stone SR, Morrison JF (1984) Catalytic mechanism of the dihydrofolate reductase reaction as determined by pH studies. Biochemistry 23:2753–2758
Viola RE, Cook PF, Cleland WW (1979) Stereoselective preperration of deuterated reduced nicotinamide adenine dinucleotide and substrates by enzymatic synthesis. Anal Biochem 96:334–340
Wang Z, Singh P, Czekster CM, Kohen A, Schramm VL (2014) Protein mass-modulated effects in the catalytic mechanism of dihydrofolate reductase: Beyond promoting vibration. J Am Chem Soc 136:8333–8341
Williams JW, Morrison JF, Duggleby RG (1979) Methotrexate, a high-affinity pseudosubstrate of dihydrofolate reductase. Biochemistry 18:2567–2573
Wright DB, Banks DD, Lohman JR, Hilsenbeck JL, Gloss LM (2002) The effects of salts on the activity and stability of Escherichia coli and Haloferax volcanii dihydrofolate reductases. J Mol Biol 323:327–344
Zusman T, Rosenshine I, Boehm G, Jaenicke R, Leskiw B, Mevarech M (1989) Dihydrofolate resuctase of the extremely halophilic archaebacterium Halobacterium volcanii. J Biol Chem 264:11878–11883
Acknowledgements
This work was supported financially by a Sasagawa Scientific Research Grant from the Japan Science Society (No. 26-315 to Y. M.) and the Platform Project for Supporting in Drug Discovery and Life Science Research (Platform for Dynamic Approaches to Living System) from Japan Agency for Medical Research and Development (AMED).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by H. Atomi.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Miyashita, Y., Ohmae, E., Ikura, T. et al. Halophilic mechanism of the enzymatic function of a moderately halophilic dihydrofolate reductase from Haloarcula japonica strain TR-1. Extremophiles 21, 591–602 (2017). https://doi.org/10.1007/s00792-017-0928-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-017-0928-0