
Abstract
Deinococcus radiodurans is highly resistant to ionizing radiation and UV radiation, and oxidative stress caused by such radiations. NADP(H) seems to be important for this resistance (Slade and Radman, Microbiol Mol Biol Rev 75:133–191; Slade, Radman, Microbiol Mol Biol Rev 75:133–191, 2011), but the mechanism underlying the generation of NADP(H) or NAD(H) in D. radiodurans has not fully been addressed. Intracellular concentrations of NAD+, NADH, NADP+, and NADPH in D. radiodurans are also not determined yet. We found that cell extracts of D. radiodurans catalyzed reduction of NAD(P)+ in vitro, indicating that D. radiodurans cells contain both enzymes and a high concentration of substrates for this activity. The enzyme and the substrate were attributed to glucose-6-phosphate dehydrogenase and glucose-6-phosphate of which intracellular concentration was extremely high. Unexpectedly, the intracellular concentration of NAD(H) was also much greater than that of NADP(H), suggesting some significant roles of NADH. These unusual features of this bacterium would shed light on a new aspect of physiology of this bacterium.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Deinococcus radiodurans, an organotrophic bacterium with a proteolytic lifestyle (Murray 1986), is remarkably resistant to oxidative stress induced by reactive oxygen species (ROS), which can be produced metabolically or form upon exposure to chemical and physical agents such as hydrogen peroxide, mitomycin C, UV radiation, ionizing radiation, and desiccation (Ghosal et al. 2005; Slade and Radman 2011). Due to its extremely high resistance to ionizing radiation, D. radiodurans has been used for investigations of radiobiology. Recent studies suggest that an extremely high ROS-scavenging capacity is responsible for this bacterium’s radiation resistance; as a result, its proteome is better protected against ROS-induced oxidative damage than those of radiation-sensitive species (Daly 2009; Daly et al. 2007; Slade and Radman 2011). Intracellular small-molecule antioxidants, including manganese, phosphate, nucleosides, and especially manganese–metabolite complexes, were recently identified as powerful ROS scavengers in D. radiodurans (Daly et al. 2010).
NADPH, the reduced form of NADP+, is involved in anabolic reactions and protects cells against oxidative stress by donating reducing equivalents to proteins such as thioredoxin and small molecules such as oxidized glutathione (GSSG) (Lu and Holmgren 2014; Obiero et al. 2010; Slade and Radman 2011). By contrast, NAD(H) is primarily involved in catabolic reactions (Kawai and Murata 2008). Given the crucial role of NADPH in resistance to oxidative stress, it is possible that NADPH protects D. radiodurans against oxidative stress and other stresses such as UV and ionizing radiation (Slade and Radman 2011). Consistent with this, the thioredoxin system of D. radiodurans is NADPH-dependent (Obiero et al. 2010).
Glucose-6-phosphate dehydrogenase (G6PDH) is the enzyme that catalyzes the first step of the pentose phosphate pathway (PPP), which generates NADPH (Stincone et al. 2015). In general, two NADPH molecules are generated from one G6P molecule via the two oxidation reactions of the PPP. Additionally, PPP generates precursors for nucleotide and amino-acid biosynthesis, such as ribose-5-phosphate and erythrose-4-phosphate (Stincone et al. 2015). Thus, the PPP is generally important for redox homeostasis and recovery from damage caused by oxidative stresses. In D. radiodurans, G6PDH appears to be critical for UV resistance: a D. radiodurans mutant lacking the G6PDH gene grows more slowly than the wild-type (WT) strain and is more sensitive to UV, H2O2, and mitomycin C (Liu et al. 2008; Zhang et al. 2003).
Despite of the significance, the mechanism underlying generation of NADP(H) or NAD(H) in D. radiodurans has not fully been addressed. Intracellular concentrations of NAD+, NADH, NADP+, and NADPH in D. radiodurans are also not determined yet. In this study, we detected the high NAD(P)+-reducing activity in the cell extracts of D. radiodurans in vitro. The activity was attributed to glucose-6-phosphate dehydrogenase and extremely high concentration of glucose-6-phosphate. We also revealed extremely high concentration of NAD(H) in this bacterium.
Materials and methods
Strains, media, and growth conditions
Bacterial strains used in this study are shown in Table 1. D. radiodurans was grown aerobically in TY medium [1% (w/v) tryptone (Nacalai, Kyoto, Japan) and 0.5% (w/v) yeast extract (Nacalai), pH 7.1] or TGY medium [0.1% (w/v) D-glucose in TY medium, pH 7.1]. E. coli was grown aerobically in TGY medium. Growth in TY or TGY medium was conducted at 32 °C and 130 strokes per minutes (spm) in a PERSONAL-11 shaker incubator (TAITEC, Koshigaya, Japan). When inoculating bacterial strains into liquid media, A 600 was adjusted to 0.1. For plasmid construction, E. coli was aerobically grown in LB medium [1% (w/v) tryptone, 0.5% (w/v) yeast extract, and 1% (w/v) NaCl] or MMI medium [2.5% (w/v) yeast extract, 1.25% (w/v) tryptone, 0.85% (w/v) NaCl, 20 mM Tris–HCl (pH 7.2), and 0.4% (v/v) glycerol] at 32 °C. Media were solidified with 1.5% (w/v) agar. If necessary, ampicillin (Ap, at 100 μg/mL) (Wako Pure Chemical Industries, Osaka, Japan) or kanamycin [Km, at 25 μg/mL (solid media) or at 10 μg/mL (liquid media)] (Wako) was added to the media.
Preparation of cell extract
D. radiodurans wild-type (WT; MK5205), G6PDH disruptant (ΔDR1596::Kmr; MK5368), and E. coli MG1655 strains were grown in 1.5 L TGY and TY media (32 °C, 100 spm) until A 600 reached 0.4–0.8. In the case of D. radiodurans, A 600 of 1.0 corresponded to 5.6 × 107 cells (colony forming units) per mL, which were determined after plating the culture onto TGY solid medium. Cells were collected by centrifugation at 6000×g for 10 min at 4 °C, washed once in 20 mM Tris–HCl (pH 7.5), and suspended in 20 mM Tris–HCl (pH 7.5) to 0.5 g wet weight per mL. The cell suspension was stored at −30 °C and thawed at room temperature. E. coli MG1655 cells were disrupted by sonication for 20 min using an INSONATOR 201 M (Kubota, Tokyo, Japan). Disruption of MK5205 and MK5368 cells was performed as follows. First, the cell suspension was dispensed into screw-cap tubes (1.5 mL/tube; 1.68 × 1010 cells/tube), and glass beads (0.1–0.2 mm diameter) were added to nearly top of the tube. Cells were disrupted by shaking the tubes for 30 s on a FastPrep24 (MP Biomedicals, Santa Ana, CA, USA) at 6.5 m/s, followed by incubation on ice for 30 s; this process was repeated a total of five times. Disrupted cells were centrifuged at 20,000×g for 10 min at 4 °C (MK5205 and MK5368) or 20 min (E. coli MG1655), and the resultant supernatant was used as cell extract. Protein concentration in the cell extract was determined by Bradford assay (Bradford 1976) using bovine serum albumin (Sigma, St. Louis, MO, USA) as a standard.
Assay of NAD(P)+-reducing activity in the cell extract
For measurement of NAD(P)+-reducing activity, A 340 of the reaction mixture [800 μL; 5 mM NAD+ or NADP+, 100 mM Tris–HCl (pH7.5), and cell extract (1 mg/mL protein)] was monitored continuously at 32 °C. Reaction was initiated by adding NAD(P)+.
For measurement of total NAD(P)+-reducing activity, the same reaction mixture [100 μL per tube] was incubated at 32 °C, and the A 340 of the supernatant was measured until saturation after tenfold dilution with 10 mM Tris (pH 11) followed by centrifugation at 20,000×g for 3 min at 4 °C. The saturated (maximum) A 340 in the 1-mL reaction mixture was regarded as the total NAD(P)+-reducing activity.
Analysis of NAD(P)+-reducing activity in the reaction mixture
Reaction mixture [300 μL; 5 mM NAD(P)+, cell extract (1 mg/mL protein), and 100 mM Tris–HCl (pH 7.5)] was incubated at 30 °C for 10 min, and 100 μL of the reaction mixture was added to 150 μL of 10 mM Tris (pH 11) (no treatment), 0.17 M NaOH (alkali treatment), or HCl (acid treatment). Alkali- or acid-treated mixtures were boiled for 90 s. Absorbance spectrum of the alkali-treated, acid-treated, and untreated mixtures were measured between the wavelengths of 200 and 400 nm (0.5 nm resolution) on a UV-1800 spectrophotometer (Shimadzu, Kyoto, Japan). Baselines were mixtures of 100 μL of 100 mM Tris–HCl (pH 7.5) and 150 μL of 10 mM Tris (pH 11), or acid- or alkali-treated 10 mM Tris (pH 11), as appropriate.
For HPLC analysis, the original reaction mixture was diluted fivefold in 10 mM Tris (pH 11) for detection of NADH. For detection of NADPH, the original reaction mixture was diluted 2.5-fold in 10 mM Tris (pH 11). After dilution, reaction mixtures were passed through a Cosmonice filter W (pore size 0.45 μm, Nacalai). The filtered mixture (10 μL) was analyzed with HPLC equipped with a COSMOSIL/COSMOGEL 5C18-AR-300 (4.6 × 150 mm; Nacalai) column. Solvent composition was changed from a 50 mM Tris–HCl (pH 7.5):methanol ratio of 100:0 to 90:10 over 40 min, and the final solvent composition was maintained for an additional 40 min (Mori et al. 2005). Signals were detected at A 340.
Disruption of the G6PDH gene in D. radiodurans
Disruption of DR_1596, the G6PDH gene of D. radiodurans (Zhang et al. 2003), was performed as described previously (Nishida and Narumi 2002) with some modifications. Details are provided in Supplementary Information.
Assay of G6PDH activity in cell extract
G6PDH activity was assayed by continuously monitoring A 340 of the reaction mixture [800 μL; 5 mM MgCl2, 5 mM G6P, 5 mM NAD(P)+, cell extract, and 100 mM Tris–HCl (pH 7.5)] at 32 °C. Protein concentration in the mixture was adjusted to 0.5 mg/mL. Reaction was initiated by adding G6P. One unit of G6PDH activity was defined as the activity that generated 1 μmol of NAD(P)H in 1 min at 32 °C. Apparent kinetic constants were determined from the Lineweaver–Burk plot.
UV resistance assay
D. radiodurans WT (MK5205) and G6PDH-disruptant (MK5368) strains were grown in 4 mL TGY or TY media until A 600 reached 0.4–0.8; A 600 of 1.0 corresponds to 5.6 × 107 cells (colony forming units) per mL as described above. Cultures were diluted in sterilized water (Zhang et al. 2003) and the cells corresponding to 2,000–4,000 cells were spread on TGY or TY agar plate (diameter = 8.5 cm). The plates were irradiated using a UV transilluminator (BIO CRAFT, Tokyo, Japan) equipped with Vilber-Lourmat T-8C, UV-C 4E (diameter = 1.5 cm; 5 lamps). The distance between the lamps and the plate was 2.5–3.0 cm. After exposure to UV, plates were incubated at 30 °C for 2–3 days. Colonies were counted, and survival rate was calculated relative to non-radiated samples.
Measurement of G6P in cell extract
Cell extracts of D. radiodurans WT (MK5205), G6PDH disruptant (MK5368), and E. coli MG1655 strains were diluted with 20 mM Tris–HCl (pH 7.5) to adjust protein concentration to 2 mg/mL, and then cleared by centrifugation at 20,000×g for 5 min at 4 °C after the diluted cell extracts were boiled for 5 min. In the case of D. radiodurans, the cell extract (approximately 1 mL containing 5.5 mg protein) was prepared. The total amount of G6P in the cell extract (G6Pt) was measured based on the increased A 340 of the mixture [total 500 μL; 1:1 of cleared supernatant: 2× reaction mixture consisting of 10 mM MgCl2, 1 mM NADP+, 200 mM Tris–HCl (pH 7.5), and 0.2 U G6PDH (Sigma)] incubated at 37 °C for 30–60 min until A 340 became stable. After boiling for 5 min in this condition, no degradation of G6P was observed. Assuming that D. radiodurans cells are spherical bodies 1 μm in diameter (Slade and Radman 2011), the volume of a single cell (4/3πr3) is 5.24 × 10− 10 μL. Thus, the total volume of 1.68 × 1010 cells per tube, from which cell extracts were prepared, was 8.80 μL. Assuming that intracellular G6P was extracted thoroughly from 1.68 × 1010 cells, the intracellular G6P concentration could be calculated by dividing G6Pt by 8.80 μL. It should be noted that the calculated G6P concentration represents a minimum value, as some cells might have remained intact.
Estimation of intracellular NAD(P)+ and NAD(P)H concentration
D. radiodurans was cultured in TGY media until A 600 reached 0.8–1.0. Cells (2.5 × 108 cells/tube) were collected by centrifugation at 15,000×g for 2 min at 4 °C, washed once with 1 mL of 20 mM Tris–HCl (pH 7.5), and immediately stored at −80 °C. Extraction of NAD(P)+ and NAD(P)H was performed as previously reported (Leonardo et al. 1996) using 125 μL/tube of 0.2 M HCl [for extraction of NAD(P)+] or 0.2 M NaOH [for extraction of NAD(P)H]. Assays of extracts containing NAD(P)+ and NAD(P)H were performed by cycling assay (details provided Supplementary Information) (Bernofsky and Swan 1973; Leonardo et al. 1996). As described above, the volume of a single cell was calculated as 5.24 × 10− 10 μL. NAD(P)+ and NAD(P)H were extracted from 2.5 × 108 cells, so the total cell volume was 0.131 μL. Assuming that intracellular NAD(P)+ and NAD(P)H were extracted thoroughly into 125 μL of extract, intracellular NAD(P)+ and NAD(P)H concentration (C in) could be calculated from that of the cell extract (C ex) using the equation: C in = (C ex × 125)/0.131.
Bioinformatics
Bioinformatic searches were conducted using The SEED Viewer (http://pubseed.theseed.org/seedviewer.cgi) (Overbeek et al. 2005) or GenomeNet (http://www.genome.jp).
Results
NAD(P)+-reducing activity in D. radiodurans cell extract
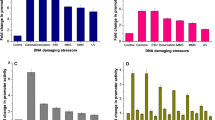
During our experiments using cell extract of D. radiodurans, we observed an obvious increase in A 340 in a simple reaction mixture consisting of the extract, 5 mM NAD+ or NADP+, and 100 mM Tris–HCl (pH 7.5) alone (Fig. 1a; TGY, black bar), suggesting that NAD(P)+ was reduced to NAD(P)H via an endogenous NAD(P)+-reducing enzymatic activity using endogenous substrates in the extract.

NAD(P)+-reducing activity in D. radiodurans cell extract. a NAD+-reducing activity (left panel) and NADP+-reducing activity (right panel) in cell extracts of D. radiodurans WT (black), D. radiodurans G6PDH disruptant (gray), and E. coli MG1655 (white) grown in TGY or TY medium. n = 3, Ave. ± SD. *p < 0.05, **p < 0.01, nd not detected. ND not determined. b Effect of the concentrations of NAD+ (closed symbols) and NADP+ (open symbols) on NAD(P)+-reducing activities in cell extracts of D. radiodurans WT (circles) and G6PDH disruptant (squares) at NAD(P)+ concentrations of 0–5 mM. n = 2, Ave. ± SD. c Total NAD(P)+-reducing activity in cell extracts of TGY-grown cells, as in a, b. n = 2, Ave. ± SD
To confirm that NAD(P)+ was reduced to NAD(P)H, we incubated the reaction mixture for 10 min and analyzed it by absorption spectroscopy and HPLC (Fig. 2a, b). Absorption spectra of the incubated reaction mixture, as well as a NaOH-treated reaction mixture, exhibited a peak near 340 nm, whereas the spectrum of HCl-treated reaction mixture did not (Fig. 2a). This observation supports the idea that NAD(P)+ is reduced to NAD(P)H, which is alkaline-stable and acid-unstable. In the HPLC analysis, NADPH was detected in the incubated reaction mixture (Fig. 2b). When the reaction mixture contained NAD+, the peak was observed at a slightly later elution time (~2 min); we attributed this to the degradation of the NADH formed in the incubated reaction mixture. Considering these results, we concluded that NAD+ and NADP+ were reduced to NADH and NADPH, respectively, when they were incubated for 10 min with cell extract of D. radiodurans and Tris–HCl (pH 7.5).

Reduction of NAD(P)+ to NAD(P)H. a UV-absorption spectrum of a mixture containing NAD+ (left panel) or NADP+ (right panel). Reaction mixture was subjected to no treatment (solid lines), alkaline treatment (dashed lines), or acid treatment (dotted lines). b Chromatograms of HPLC analysis. Upper panel 0.2 mM NADH (gray line), mixture consisting of 5 mM NAD+ and D. radiodurans cell extract (black line). Lower panel 0.2 mM NADPH (gray line), mixture consisting of 5 mM NADP+ and D. radiodurans cell extract (black line). Arrows indicate peaks corresponding to the generated NAD(P)H
The NAD(P)+-reducing activity was saturated in the presence of >2.5 mM NAD(P)+, and exhibited higher affinity for NAD+ than for NADP+ (Fig. 1b; Supplementary Fig. S1). Moreover, NAD+-reducing activity was higher than NADP+-reducing activity (Fig. 1a, b; Supplementary Fig. S1). The total NAD(P)+-reducing activity was calculated as 3.5–4.0 mg−1 (Fig. 1c; black bar). By contrast, when cell extract of TGY-grown E. coli was used, we detected no NAD+-reducing activity and only a little NADP+-reducing activity (Fig. 1a; white bar). The total NAD(P)+-reducing activity of E. coli cell extract was also much lower than that of TGY-grown D. radiodurans (Fig. 1c; white bar).
In TGY-grown D. radiodurans, the intracellular concentrations of NAD+, NADH, and NADPH were calculated to be 6.5 ± 0.8 mM (NAD+), 37 ± 1 mM (NADH), and 0.72 ± 0.39 mM (NADPH), respectively, whereas NADP+ was not detectable (Fig. 3; black bar, Table 2).

Intracellular concentrations of NAD(P)+ and NAD(P)H in TGY-grown D. radiodurans. Intracellular concentrations of NAD(H) (left panel) and NADP(H) (right panel) of D. radiodurans WT (black bars) and G6PDH disruptant (gray bars). n = 3, Ave. ± SD, **p < 0.01, nd not detected
Contribution of G6P and G6PDH to NAD(P)+-reducing activity
Taking into account previous observations that G6PDH plays a critical role in survival of D. radiodurans under oxidative stress (Liu et al. 2008; Zhang et al. 2003), we sought to understand how this enzyme is involved in NAD(P)H generation. To this end, we constructed a D. radiodurans G6PDH disruptant (MK5368). Cell extract from TGY-grown D. radiodurans WT strain exhibited NADP+- and NAD+-dependent G6PDH activity, with higher affinity for NADP+ (apparent K m: NADP+, 12 μM; NAD+, 53 μM; apparent V max: NADP+, 0.032 U/mg; NAD+, 0.053 U/mg) (Supplementary Fig. S2). No NADP+-/NAD+-dependent G6PDH activity was detectable in cell extract from the D. radiodurans G6PDH disruptant (data not shown). In agreement with previous reports (Liu et al. 2008; Zhang et al. 2003), the D. radiodurans G6PDH mutant grew more slowly than the WT strain and was more sensitive to UV (Fig. 4). Next, we examined the NAD(P)+-reducing activity of this disruptant (Fig. 1; TGY, gray bar). Notably, very little of NADP+-reducing activity was detected in the mutant, whereas the NAD+-reducing activity was approximately 50% of that of the WT strain (Fig. 1a, b; Supplementary Fig. S1). These data indicated that NAD(P)+-reducing activity, that was observed in vitro, was attributed to G6PDH.

Contribution of G6PDH and G6P. a Growth of D. radiodurans WT (closed circles, n = 3) and G6PDH disruptant (closed squares, n = 2) in TGY media. Ave. ± SD. b Sensitivity to UV of D. radiodurans WT (closed symbols) and G6PDH disruptant (open symbols) grown in TGY (circles) or TY (squares) media. Survival rate was defined as the ratio of colony count after UV exposure to the count in non-UV-irradiated samples. Ave. ± SD, n = 3, *p < 0.05 (vs. WT in TGY medium), † p < 0.01 (vs. WT in TY medium)
Thus, we expected that G6P would be present at a high concentration in D. radiodurans cells. Accordingly, an intracellular G6P concentration was 106 mM (173 μmol/g protein) (Fig. 5; black bar, Table 3). In cleared cell extract from TGY-grown D. radiodurans G6PDH disruptant, approximately the same level of G6P was detected (Fig. 5). TGY-grown E. coli was treated using the same method, but no G6P was detected in the cleared cell extract (Fig. 5; black bar).

Intracellular G6P. Intracellular G6P levels of D. radiodurans (Dra) WT, G6PDH disruptant (Δg6pdh), and E. coli MG1655 (Eco) grown in TGY (black bars) or TY (white bars) medium (n = 3–5, Ave. ± SD). Values are shown as μmol of G6P per 1 g of cellular protein. nd not detected
Intracellular concentrations of NAD+, NADH, NADP+, and NADPH in the TGY-grown D. radiodurans G6PDH disruptant were calculated as 6.1 ± 2.6 mM (NAD+), 11 ± 2 mM (NADH), 0.084 ± 0.118 mM (NADP+), and 0.26 ± 0.07 mM (NADPH) (Fig. 3, gray bar; Table 2).
Effect of glucose on NAD(P)+-reducing activity and concentration of G6P
We examined the effect of glucose in the medium on NAD(P)+-reducing activity, G6P content, and UV sensitivity (Figs. 1a, 4, 5; white bar). Glucose had no effect on growth (data not shown) or UV sensitivity (Fig. 4b), but unexpectedly, NAD(P)+-reducing activity was higher in extract of TY-grown cells (without glucose) than in extract of TGY-grown cells (with glucose) (Fig. 1a).
Discussion
In this study, we observed an increase in the A 340 of D. radiodurans cell extract when NAD(P)+ was added, which was due to NAD(P)+ reduction by endogenous enzymes and substrates. The enzyme and substrate responsible for the observed reducing activity was attributed to G6PDH and G6P in D. radiodurans cells (intracellular concentration of G6P was 106 mM, corresponding to 173 μmol/g protein). Compared with those of other microorganisms (E. coli, Saccharomyces cerevisiae, Mycobacterium smegmatis, Corynebacterium glutamicum), this value of D. radiodurans is extremely high (Table 3).
To what extent G6P is synthesized in D. radiodurans cells? Although G6P can be synthesized by the PTS-dependent glucose uptake system (Deutscher et al. 2006), no genes encoding components of this system have been detected in the genome of D. radiodurans (The SEED Viewer and GenomeNet). G6P is also synthesized from glucose by glucokinase (GK) (Kawai et al. 2005); two GK genes are present in the D. radiodurans genome (DR_0823 and DR_2296). However, in D. radiodurans grown without glucose, the intracellular G6P level was not significantly different from that of D. radiodurans grown with glucose (Fig. 5), indicating that GK is not responsible for the production of G6P in this bacterium. D. radiodurans utilizes protein as its main carbon source (Slade and Radman 2011), and glucose is a less optimal carbon source than amino acids and fructose (Slade and Radman 2011; Venkateswaran et al. 2000). The D. radiodurans genome contains genes encoding all enzymes of gluconeogenesis. Taking these facts into consideration, it is likely that D. radiodurans synthesizes G6P via gluconeogenesis rather than GK or the PTS system.
Generally, NADP+ is biosynthesized from NAD+ by NAD kinase (NADK) (Kawai and Murata 2008). However, no homologue of NADK gene was found in the genome of D. radiodurans using The SEED Viewer and GenomeNet. Also, intracellular concentrations of NADP+ and NADPH, as well as NAD+ and NADH in D. radiodurans had also not been determined yet. In this study, for the first time, we detected NADPH in D. radiodurans WT and NADP+ in the G6PDH disruptant (Fig. 3; Table 2). This result indicates that D. radiodurans is able to synthesize NAD(P)H. The reason why NADP+ was undetectable in cell extract of D. radiodurans WT (Fig. 3; Table 2) was speculated that intracellular NADP+ would be reduced into NADPH to the level below detection.
In this study, we found that D. radiodurans cells contain a high concentration (37 mM) of NADH (Fig. 3; Table 2), possibly consistent with the observation that the NAD+-reducing activity was higher than the NADP+-reducing activity (Fig. 1a). Generally, NADH and NAD+ engage in catabolic reactions, and the intracellular concentration of NAD+ tends to exceed that of NADH; the intracellular NAD+/NADH ratio is on the order of 700 to 1 (Ying 2008). However, the intracellular concentration of NADH in D. radiodurans that we measured in this study is higher than those in other species (Table 2); consequently, the intracellular NAD+/NADH ratio was much lower (Table 4), suggesting some significant roles of NADH in D. radiodurans. These unusual features of this bacterium would shed light on a new aspect of the physiology of D. radiodurans.
The D. radiodurans G6PDH disruptant grew more slowly than the WT strain and was more sensitive to UV, as reported previously (Liu et al. 2008; Zhang et al. 2003). Zhang et al. reported the survival index (% survival of mutant/% survival of WT) of the D. radiodurans G6PDH disruptant after exposure to UV were 0.16–0.49 (Zhang et al. 2003), that is similar to the survival index (0.17) of the G6PDH disruptants after exposure to UV for 15 min (Fig. 4). Zhang et al. attributed the UV-sensitivity of the G6PDH disruptant to the low levels of UMP and IMP that are demanded for excision repair to repair its DNA (Zhang et al. 2003). Our data suggest that the high concentration of G6P in D. radiodurans would guarantee a good supply of substrates and reducing power such as NADH demanded for several cellular processes including the protection from UV-damage.
Abbreviations
- ROS:
-
Reactive oxygen species
- GSSG:
-
Oxidized glutathione
- G6PDH:
-
Glucose-6-phosphate dehydrogenase
- G6P:
-
Glucose-6-phosphate
- WT:
-
Wild type
- GK:
-
Glucokinase
- NADK:
-
NAD kinase
References
Bernofsky C, Swan M (1973) An improved cycling assay for nicotinamide adenine dinucleotide. Anal Biochem 53:452–458
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem 72:248–254
Daly MJ (2009) A new perspective on radiation resistance based on Deinococcus radiodurans. Nat Rev Microbiol 7:237–245
Daly MJ et al (2007) Protein oxidation implicated as the primary determinant of bacterial radioresistance. PLoS Biol 5:e92
Daly MJ et al (2010) Small-molecule antioxidant proteome-shields in Deinococcus radiodurans. PLoS One 5:e12570
Deutscher J, Francke C, Postma PW (2006) How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol Mol Biol Rev 70:939–1031
Dominguez H et al (1998) Carbon-flux distribution in the central metabolic pathways of Corynebacterium glutamicum during growth on fructose. Eur J Biochem 254:96–102
dos Santos MM et al (2004) Manipulation of malic enzyme in Saccharomyces cerevisiae for increasing NADPH production capacity aerobically in different cellular compartments. Metab Eng 6:352–363
Gao H et al (2007) Altered poly(ADP-ribose) metabolism impairs cellular responses to genotoxic stress in a hypomorphic mutant of poly(ADP-ribose) glycohydrolase. Exp Cell Res 313:984–996
Ghosal D et al (2005) How radiation kills cells: survival of Deinococcus radiodurans and Shewanella oneidensis under oxidative stress. FEMS Microbiol Rev 29:361–375
Grose JH, Joss L, Velick SF, Roth JR (2006) Evidence that feedback inhibition of NAD kinase controls responses to oxidative stress. Proc Natl Acad Sci USA 103:7601–7606
Hasan MR et al (2010) Glucose 6-phosphate accumulation in mycobacteria: implications for a novel F420-dependent anti-oxidant defense system. J Biol Chem 285:19135–19144
Kawai S, Murata K (2008) Structure and function of NAD kinase and NADP phosphatase: key enzymes that regulate the intracellular balance of NAD(H) and NADP(H). Biosci Biotechnol Biochem 72:919–930
Kawai S et al (2001) Molecular characterization of Escherichia coli NAD kinase. Eur J Biochem 268:4359–4365
Kawai S et al (2005) Hypothesis: structures, evolution, and ancestor of glucose kinases in the hexokinase family. J Biosci Bioeng 99:320–330
Leonardo MR, Dailly Y, Clark DP (1996) Role of NAD in regulating the adhE gene of Escherichia coli. J Bacteriol 178:6013–6018
Liu X et al (2008) Resistance of Deinococcus radiodurans to mutagenesis is facilitated by pentose phosphate pathway in the mutS1 mutant background. Curr Microbiol 57:66–71
Lu J, Holmgren A (2014) The thioredoxin antioxidant system. Free Radical Biol Med 66: 75–87
Lundquist R, Olivera BM (1971) Pyridine nucleotide metabolism in Escherichia coli. I. Exponential growth. J Biol Chem 246:1107–1116
Mori S et al (2005) Molecular conversion of NAD kinase to NADH kinase through single amino acid residue substitution. J Biol Chem 280:24104–24112
Murray RG (1986) Family II. Deinococcaceae. In: Sneath PHA, Mair NS, Sharpe ME, Holt JG (eds) Bergey’s manual of systematic bacteriology. Williams & Wilkins Co., Baltimore, MD
Nishida H, Narumi I (2002) Disruption analysis of DR1420 and/or DR1758 in the extremely radioresistant bacterium Deinococcus radiodurans. Microbiology 148:2911–2914
Obiero J, Pittet V, Bonderoff SA, Sanders DA (2010) Thioredoxin system from Deinococcus radiodurans. J Bacteriol 192:494–501
Overbeek R et al (2005) The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res 33:5691–5702
Slade D, Radman M (2011) Oxidative stress resistance in Deinococcus radiodurans. Microbiol Mol Biol Rev 75:133–191
Sporty J et al (2009) Quantitation of NAD+ biosynthesis from the salvage pathway in Saccharomyces cerevisiae. Yeast 26:363–369
Stincone A et al (2015) The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc 90:927–963
Theobald U et al (1997) In vivo analysis of metabolic dynamics in Saccharomyces cerevisiae : I. Experimental observations. Biotechnol Bioeng 55:305–316
Venkateswaran A et al (2000) Physiologic determinants of radiation resistance in Deinococcus radiodurans. Appl Environ Microbiol 66:2620–2626
Ying W (2008) NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal 10:179–206
Zhang YM, Liu JK, Wong TY (2003) The DNA excision repair system of the highly radioresistant bacterium Deinococcus radiodurans is facilitated by the pentose phosphate pathway. Mol Microbiol 48:1317–1323
Acknowledgements
This work is supported by the JSPS KAKENHI Grant Number 15K07387.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. da Costa.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yamashiro, T., Murata, K. & Kawai, S. Extremely high intracellular concentration of glucose-6-phosphate and NAD(H) in Deinococcus radiodurans . Extremophiles 21, 399–407 (2017). https://doi.org/10.1007/s00792-016-0913-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-016-0913-z