
Abstract
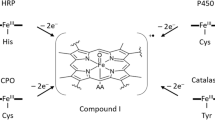
Oxoiron(IV) porphyrin π-cation-radical complexes (Cpd I) have been studied as models for reactive intermediates called compound I in cytochromes P450, peroxidases, and catalases. It has been well known that the electronic structure and reactivity of Cpd I are modulated by the substituted position and the electron-withdrawing ability of the substituent. However, there still remain two major questions: (1) how many electronegative halogen atoms should be introduced in the meso-phenyl group to switch the porphyrin π-cation-radical state of Cpd I? (2) How does the electron-withdrawing effect of the substituent modulate the reactivity of Cpd I? To answer these two questions, we here performed experimental and theoretical studies on the electron-withdrawing effect of the meso-substituent. We gradually increased the electron-withdrawing effect by increasing the number of fluorine atoms in the meso-phenyl group. Spectroscopic analyses of these Cpd I models reveal that the porphyrin radical state shifts from having a2u radical character to having a1u radical character with an increase in the number of the fluorine atoms in the phenyl group, and the ground state of Cpd I switches from the a2u state to the a1u state when four fluorine atoms are introduced in the meso-phenyl group. The switch of the radical state is predicted well by LC-BLYP, but not by the commonly used B3LYP. The theoretical calculations indicate that the electron-withdrawing substituent makes Cpd I more reactive by stabilizing the ferric porphyrin state (product state) more than the Cpd I state (reactant state), generating a larger free energy change in the oxygenation reaction (ΔG) of Cpd I.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
High-valent metal-oxo species have been identified as reactive intermediates in many metalloenzymes and in catalytic oxygenation reactions with transition metal complexes [1,2,3,4,5,6]. For example, in the reactions of peroxidases, catalases, and cytochrome P450, oxoiron(IV) porphyrin π-cation-radical species, which are two electron equivalents more oxidized than the ferric resting states of these enzymes, have been characterized as reactive intermediates and called Compound I [1,2,3]. Because of its biological significance and extremely high reactivity, the electronic structure and reactivity of Compound I have been studied with synthetic model complexes. Groves et al. reported the first example of an oxoiron(IV) porphyrin π-cation-radical complex (Cpd I), a synthetic model complex of Compound I, from the reaction of the iron(III) meso-tetramesitylporphyrin (TMP) complex with m-chloroperoxybenzoic acid (mCPBA) [7]. Since then, various types of Cpd Is have been prepared with various iron porphyrin complexes and oxidants [8,9,10,11,12,13,14,15,16,17,18,19]. Catalytic oxidation studies reported that iron porphyrins having electron-deficient substituents, such as 2,6-dichlorophenyl and pentafluorophenyl groups at the meso-positions, are efficient catalysts for alkane and alkene oxygenation reactions [20,21,22,23,24,25]. Cpd I of the tetrakis-2,6-dichlorophenylporphyrin was first prepared by the oxidations of the corresponding ferric perchlorate complex with mCPBA, pentafluoroiodosylbenzene, on ozone by Sawyer et al [9]. Cpd I of the tetrakis-pentafluorophenylporphyrin was first prepared by the oxidations of the corresponding ferric perchlorate complex with mCPBA by us [14, 15]. Cpd Is were also prepared from more biomimetic porphyrins, which have sterically hindered aryl groups at the pyrrole-β positions [16, 17]. The overall structure of the pyrrole-β substituted porphyrins resembles that of protoporphyrinIX used in many heme proteins.
The electronic structure and reactivity of Cpd Is have been studied with various spectroscopic methods by many groups [7,8,9,10,11,12,13,14,15,16,17,18,19, 26,27,28,29,30,31,32]. To date, there are two known types of porphyrin π-cation-radical states[16, 17], with the porphyrin π-cation radical having an unpaired electron in the a1u orbital or in the a2u orbital (Fig. 1). The identity of the porphyrin radical state of Cpd I is modulated by the substituted position of the substituent and its electron-withdrawing ability. The Cpd I of TMP, having an electron-donating mesityl group at the meso-position, has been shown to have an unpaired electron in the a2u orbital: the a2u radical state [7]. However, the Cpd I bearing very strong electron-withdrawing pentafluorophenyl groups at the meso-positions has been known to be in the a1u radical state [14, 15]. On the other hand, both Cpd Is having mesityl and pentafluorophenyl groups at the pyrrole-β positions have been reported to be in the a1u radical states [16, 17]. A detailed study revealed that the electron-withdrawing effect of the meso-substituent stabilizes the a2u orbital relative to the a1u orbital, while that of the pyrrole-β substituent stabilizes both the a1u and a2u orbitals [33]. The electron-withdrawing effect of the substituent also modulates the reactivity of Cpd Is. Competitive cyclohexene epoxidation reactions of Cpd Is showed that, as the electron-withdrawing effect of the substituent at the meso-position and the pyrrole-β position becomes stronger, Cpd I becomes more reactive [16, 17]. Recently, these results were further confirmed by detailed kinetic analysis [34].

Spin density distribution of the a1u orbital (left) and the a2u orbital (right)
The above-mentioned results on the electron-withdrawing effect of the substituent leave the following two questions: (1) how many electronegative halogen atoms should be introduced in the meso-phenyl group to switch the porphyrin π-cation-radical state of Cpd I? (2) How does the electron-withdrawing effect of the substituent modulate the reactivity of Cpd I? To answer these two questions, we here conducted an experimental and theoretical study on the electron-withdrawing effect of the meso-substituent. To shed light on the question (1), we gradually increased the electron-withdrawing effect of the substituent by increasing the number of fluorine atoms in the meso-phenyl group (Fig. 2). Spectroscopic analysis of these Cpd Is reveals that the identity of the porphyrin radical state shifts from the a2u radical character to the a1u radical character by increasing the number of the fluorine atoms in the phenyl group, and the ground state of Cpd I switches from the a2u state to the a1u state when four fluorine atoms are introduced in the meso-phenyl group. Theoretical calculations indicate that the electron-withdrawing substituent makes the Cpd I more reactive by stabilizing the ferric porphyrin state (product state) more than the Cpd I state (reactant state), hereby generating a larger free energy change of the reaction (∆G) for the oxygenation reaction of Cpd I.

Structures of 1-Cpd I–5-Cpd I used in this study
Experimental section
Instrumentation
UV–visible absorption spectra were recorded on an Agilent 8453 spectrometer (Agilent Technologies) equipped with a USP-203 low-temperature chamber (UNISOKU). 1H NMR spectra were measured on a JNM-ECA600 and Lambda-400 spectrometer (JEOL). The chemical shifts were referenced to tetramethylsilane (TMS) using the residual peaks of the deuterated solvents, chloroform (7.24 ppm) and toluene-CH3 (2.09 ppm). The concentrations of the NMR samples were 1–2 mM. EPR spectra were recorded at 4 K on Bruker E500 X-band spectrometer with a single mode cavity connected an Oxford Instrument EPR910 helium-flow cryostat (Bruker). The concentration of EPR samples was ~ 2 mM. Measurements were carried out under the following conditions: microwave frequency, 9.657 GHz; microwave power, 1.003 mW; modulation amplitude, 10 G; time constant, 82 ms. Ozone gas was generated by the UV irradiation of oxygen gas (99.999%) with an ozone generator PR-1300 (Clear Water) and used without further purification.
Materials
Anhydrous organic solvents were obtained commercially and stored in a glove box. Dichloromethane was purified by passing through an alumina column just before use in the glove box. Other chemicals were purchased commercially and used without further purification.
meso-Tetraarylporphyrins
The meso-tetraarylporphyrins (1–5) were prepared from pyrrole and the corresponding benzaldehydes according to a previously published procedure [35]. The porphyrins were purified by silica gel column with dichloromethane as the eluent. Yield: 1: 11%, 2: 35%, 3: 56%, 4: 20%, 5: 10%. Spectroscopic data. UV–Vis (nm) in dichloromethane. 1: 416, 510, 542, 586, 640. 2: 413, 508, 536, 584, 637. 3: 412, 507, 536, 584, 637. 4: 412, 506, 536, 583, 636. 5: 412, 506, 583, 635. 1H NMR (400 MHz, ppm from TMS) in CDCl3 at 298 K: 1: 8.84 (py-H), 6.92 and 6.90 (m-H), 4.06 (p-OCH3), − 2.78 (NH). 2: 8.85 (py-H), 7.78 (p-H), 7.36 (m-H), − 2.78 (NH). 3: 8.87 (py-H), 7.15 (m-H), − 2.86 (NH). 4: 8.90 (py-H), 7.62 (m-H), − 2.89 (NH). 5: 8.89 (py-H), − 2.93 (NH).
Ferric chloride complexes
The ferric chloride complexes of 1–5 were prepared by the insertion of iron into the free base porphyrins of 1–5 with ferrous chloride and sodium acetate in acetic acid. General procedure: acetic acid (100 mL) and sodium acetate (600 mg) were placed in a 300 mL of two-neck eggplant sharp flask and degassed by bubbling argon gas for 30 min. Ferrous chloride tetrahydrate (600 mg) was added to the solution under anaerobic conditions. The mixture was heated at 40 °C under the continuous bubbling of argon gas and then the free base of the porphyrin (1–5) (200 mg), dissolved in chloroform (70 mL), was added dropwise to the mixture. The mixture was heated at 60 °C for 3 h under an argon atmosphere. The progress of the reaction was monitored by TLC. After confirming the progress of the reaction, the reaction mixture was cooled to room temperature and poured into water (300 mL). The iron porphyrin was extracted three times with dichloromethane (150 mL × 3). The extracted dichloromethane solution was washed several times with water (150 mL), then a saturated sodium hydrogen carbonate solution, and then an HCl solution. After evaporation of the solvent, the remaining iron porphyrin was purified using a silica gel column and dichloromethane, then a dichloromethane–methanol mixture as eluents. The purified iron porphyrin solution was washed with water, a sodium hydroxide solution (1 M), and then an HCl solution (3 M). Yields: 1: 81%, 2: 53%, 3: 30%, 4: 65%, 5: 71%. Spectroscopic data. UV–Vis (nm) in dichloromethane. 1: 374, 414, 507, 579, 643. 2: 368, 412, 505, 578, 639. 3: 363, 412, 504, 578, 635. 4: 353, 411, 504, 630. 5: 352, 411, 504, 630. 1H NMR (400 MHz, ppm from TMS) in CDCl3 at 298 K: 1: 79.91 (py-H), 13.13 and 11.88 (m-H), 4.93(p-OCH3). 2: 81.54 (py-H), 13.8 and 12.5 (m-H), 7.42 (p-H). 3: 80.0 (py-H), 13.11 and 11.88 (m-H). 4: 80.6 (py-H), 7.35(p-H). 5: 80.6 (py-H).
Ferric trifluoroacetate complexes [36]
The ferric trifluoroacetate complexes of 1–5 were synthesized from reaction of ferric hydroxide complexes, which were prepared from the reactions of ferric chloride complexes of 1–5 with sodium hydroxide solution (1 M), with 2 equiv of trifluoroacetic acid, respectively, and purified by recrystallization from dichlorometane–hexane. Spectroscopic data. UV–Vis (nm) in dichloromethane. 1: 411, 507, 575, 640. 2: 407, 506, 577, 639. 3: 407, 506, 576, 637. 4: 409, 506, 573, 633. 5: 406, 506, 633. 1H NMR (400 MHz, ppm from TMS) in CDCl3 at 298 K: 1: 75.6 (py-H), 12.6 and 11.6 (m-H), 4.96 (p-OCH3). 2: 75.7 (py-H), 13.2 and 12.0 (m-H), 7.76 (p-H). 3: 76.2 (py-H), 12.7 and 11.6 (m-H). 4: 76.2 (py-H), 7.64 (p-H). 5: 76.3 (py-H).
Preparation of Cpd I
The ferric porphyrin complex (100 µM) in dichloromethane was prepared in a 1 cm quartz cuvette in a low-temperature chamber set on a UV–visible absorption spectrometer. After stabilization of the temperature of the solution at − 80 °C, O3 gas was slowly bubbled into the solution with a gas-tight syringe. The oxidation process was monitored using the absorption spectrometer. After confirming the generation of the oxoiron(IV) porphyrin π-cation-radical complex, the excess O3 gas in the cell was removed by bubbling argon gas with a gas-tight syringe.
NMR and EPR samples were prepared in sample tubes by the oxidations of ferric porphyrin trifluoroacetate complexes with mCPBA. Ferric porphyrin complex (2 mM) was dissolved in dichloromethane (dichloromethane-d2 for NMR) and transferred into a sample tube. The sample was cooled at − 60 °C and 4 equiv. of mCPBA in dichloromethane (dichloromethane-d2 for NMR) was added. The sample color changed from brown to green.
DFT calculations
The DFT, TD-DFT, and Hartree–Fock calculations were performed using the Gaussion09 program package [37]. B3LYP was used for molecular geometry optimization in the quartet ground state and potential energies along the reaction pathway. LC-BLYP and Hartree–Fock were additionally used for comparison of stability between the a1u and a2u radical states. For basis sets, we used cc-pVTZ set for Fe, Cl, O, N, and 6-31G(d) for C of porphyrin ring and VDZ for the other atoms.
Results and discussion
Spectroscopic characterization of Cpd Is
The ferric trifluoroacetate complexes of 1–5 were oxidized to the corresponding Cpd Is of 1–5, 1-Cpd I–5-Cpd I, by ozone gas or m-chloroperoxybenzoic acid (mCPBA) in dichloromethane at low temperature (− 60 ~ − 80 °C). To investigate the a1u/a2u porphyrin π-cation-radical character, 1-Cpd I–5-Cpd I were characterized using absorption, 1H NMR and EPR spectroscopy. The spectroscopic data, as well as the redox potentials, for 1-Cpd I–5-Cpd I are summarized in Table 1. It has been known that the absorption spectra of Cpd Is show strong absorption peaks resulting from π-cation-radical character at around 650–700 nm. Figure 3 shows absorption spectra of 1-Cpd I–5-Cpd I. These absorption spectra having strong absorption peaks near 680 nm are very close to those of Cpd Is reported previously [7,8,9,10,11,12,13,14,15,16,17,18,19]. The absorption spectral changes have clear isosbestic points in the oxidation processes, indicating almost quantitative formation of 1-Cpd I–5-Cpd I. Figure 4 shows 1H NMR and EPR spectra of 1-Cpd I–5-Cpd I. The paramagnetically shifted 1H NMR signals in the upfield and downfield regions are assignable to the pyrrole-β proton signals and the m-proton signals of the meso-phenyl groups, respectively, on the basis of the 1H NMR shifts of Cpd Is reported previously [9, 14,15,16,17]. As the electron-withdrawing effect of the meso-phenyl group becomes stronger, the paramagnetic shift of the pyrrole-β proton signal becomes larger, while that of the m-proton signal becomes smaller. These changes can be interpreted by the change in the a1u/a2u porphyrin π-cation-radical character and this is discussed in the next section.

Absorption spectral change for the oxidation of iron(III) porphyrin trifluoroacetate complexes of 1–5 with ozone gas in dichloromethane at − 80 °C. (a) 1, (b) 2, (c) 3, (d) 4, (e) 5. Black line: iron(III) porphyrin trifluoroacetate complex, green line: Cpd I, gray line: in the oxidation

1H NMR (left) spectra in dichloromethane-d2 at –60 °C and EPR (right) spectra in dichloromethane at 3.8 K for 1-Cpd I–5-Cpd I. The intense peaks in the diamagnetic region result from solvent and mCPBA used for the preparation. The sharp signals around 1000 gauss in EPR spectra are due to unoxidized iron(III) trifluoroacetate complexes
EPR spectra of 1-Cpd I–5-Cpd I show three signals with gx and gy ~ 4 and gz ~ 2 at 3.8 K. These EPR spectra are consistent within a quartet (S = 3/2) ground state, confirming the oxoiron(IV) porphyrin π-cation-radical states for 1-Cpd I–5-Cpd I due to ferromagnetic coupling of iron(IV) spins (S = 1) with porphyrin π-cation-radical spin (S = 1/2). As the electron-withdrawing effect of the meso-phenyl group is stronger, the gx signal shifts to the gy signal and the g-anisotropy becomes smaller. The change of the g value can be interpreted by the change of the strength of the magnetic interaction (J/D value) between the ferryl iron spin and the porphyrin π-cation-radical spin [26], which is caused by the change in the a1u/a2u porphyrin π-cation-radical character. Details are discussed in the next section.
The EPR spectrum for 5-Cpd I resembles an axial spectrum (gx = gy) and close to that of Compound I of ascorbate peroxidase (ASP-I) [38]. The present EPR spectrum for 5-Cpd I is different from that previously prepared from ferric perchlorate complex, which affords the EPR signal around g = 2 [14]. As shown in the EPR spectra of compounds I of various heme enzymes [33, 38], the axial ligand changes the EPR spectrum of compound I by changing the spin interaction. Thus, the change would be due to the difference in the axial ligands: perchlorate and trifluoroacetate. In addition, the similarity of the EPR spectra between 5-Cpd I and ASP-I suggests the a1u radical state for ASP-I.
Switching of the a 1u/a 2u porphyrin π-cation-radical state
The 1H NMR and EPR data of the high-valent complexes are reasonably interpreted by the change in the porphyrin π-cation-radical character from the a2u radical state to the a1u radical state by increasing the electron-withdrawing effect of the meso-phenyl group. As shown in Fig. 1, the meso-position has a large spin density in the a2u orbital, but is at a node in the a1u orbital. Therefore, the 1H NMR signal of the m-proton of the meso-phenyl group shows a large paramagnetic shift when Cpd I is in the a2u radical state, but that affords a very small paramagnetic shift when it is in the a1u radical state. On the other hand, both the a1u and a2u orbitals have spin density at the pyrrole-β position, but the spin density in the a1u orbital is larger than that in the a2u orbital. Thus, the paramagnetic shift of the pyrrole-β proton in the a1u radical state would be larger than that of the a2u radical state. In fact, a previous 1H NMR study reported that Cpd I of TMP affords the m-proton signal at 58.7 and 59.7 ppm and the pyrrole-β proton signal at − 13.5 ppm at − 60 °C [39]. These NMR shifts are quite reasonable for the a2u porphyrin radical state for Cpd I of TMP. 1-Cpd I shows the m-proton signals at 32.6 and 32.9 ppm and the pyrrole-β proton signal at − 54.1 ppm, which are, respectively, smaller and larger than those of the Cpd I of TMP, respectively. Previously, the pyrrole-β proton signal of 5-Cpd I, prepared from the ferric perchlorate complex of 5, was observed at − 96 ppm at − 80 °C [14]. Taking into account the difference in temperature and axial ligand, the present NMR shift (− 72.6 ppm at − 60 °C) for 5-Cpd I is comparable to the shift in the previous study. These changes are consistent with an idea that 1-Cpd I has a a2u ground state, but the a1u radical state is mixed into to the ground a2u state. As we proposed previously, the energy gap between the a2u radical state and the a1u radical state would change to be small by the electron-withdrawing effect of the meso-substituent. The 1H NMR shifts for 1-Cpd I–5-Cpd I indicate that, as the electron-withdrawing effect of the meso-phenyl group is stronger, the a1u radical character becomes stronger. The drastic change in the pyrrole-β proton NMR shift can be found between 3-Cpd I and 4-Cpd I, indicating that the ground radical state switches from the a2uradical state to the a1u radical state at the boundary between 3-Cpd I and 4-Cpd I. The changes in the relative energies of the a2u radical state and the a1u radical state are summarized in Fig. 5. These changes can be explained by the spin density distribution in the a1u and a2u orbitals. Since the spin density at the meso-position is large in the a2u orbital, but at a node in the a1u orbital, the electron-withdrawing substituent at the meso-position stabilizes the a2u orbital more than the a1u orbital, changing from the a2u radical state to the a1u radical state.

Switch of the ground state of Cpd I by the electron-withdrawing effect of the meso-substituent, estimated from their paramagnetic shifts of Cpd I of TMP and 1-Cpd I–5-Cpd I. The energies of the excited a1u and a2u radical states are uncertain from the present experiments
EPR data also support the above discussion. The EPR g values for 1-Cpd I–5-Cpd I indicate an S = 3/2 ground spin states due to ferromagnetic interaction. The EPR spectra for 1-Cpd I and 5-Cpd I are close to those for Cpd I having the radical spin in the a2u and a1u orbitals, respectively [26]. The shift of the gx value suggests the change from the a2u radical state to the a1u radical state with increasing in the electron-withdrawing effect. Previous calculations have shown that the EPR g⊥ value becomes smaller as the ferromagnetic interaction becomes weaker [26, 33]. Therefore, the EPR g values indicate that the magnetic interaction between the ferryl iron and porphyrin π-cation-radical spins becomes weaker as the a1u radical character grows stronger. The pyrrole N atom position is at the node, but the pyrrole-α and -β positions have large spin density in the a1u orbital (Fig. 1). On the other hand, large spin density can be found at the pyrrole N and the meso-positions in the a2u orbital (Fig. 1). Therefore, the distance between the porphyrin π-radical spin and the ferryl iron spin in the a1u orbital is longer than that in the a2u orbital, resulting in the weak magnetic interaction for the a1u radical complex.
Previously, we reported the effect of the axial ligand on the a1u/a2u porphyrin π-cation-radical state of Cpd I [18]. We prepared Cpd I having various inorganic anionic axial ligands, such as chloride, nitrate, trifluoroacetate, acetate, benzoate, fluoride, etc., and biomimetic axial ligands, such as imidazoles and phenolates [19, 39]. These studies showed that the a1u/a2u porphyrin π-cation-radical state is not switched by the identity of the axial ligand and it is determined by the identity of the porphyrin ligand. However, it still remains unclear for the effect of thiolate axial ligand, because Cpd I having thiolate axial ligand has not been prepared.
DFT calculations
To confirm the electron-withdrawing effect of the porphyrin ligand, we performed the DFT calculations on 2-Cpd I–5-Cpd I, as well as Cpd I of meso-tetraphenylporphyrin (F0), F0-Cpd I (Fig. 6). Chloride is commonly used as the axial ligands of the model complexes to simplify the calculations. The optimized structures are shown in Figure S1. The structural and electronic parameters are summarized in Tables S1–S3. The energies of the ground state and the excited states are summarized in Table S4. Ionization potentials (IP) for oxoiron(IV) porphyrin complexes of 2–5 are listed in Table 1 to compare their redox potentials. The calculated ionization potentials showed linear correlation with their redox potentials (Figure S2). Although the molecular symmetry of the model complexes is reduced from D4h to C4v or C4 by the coordination of the O=Fe–Cl moiety and the tilt of the meso-phenyl groups, we here denote here the porphyrin orbital containing the π-cation radical with the symmetry label from D4h symmetry: the a1u orbital or the a2u orbital (Fig. 1). The ground spin states of all the Cpd Is are the quartet states, in which the ferryl iron spins (S = 1) ferromagnetically couple with the porphyrin π-cation-radical spin (S = 1/2). However, the Mulliken spin populations on the meso-C and pyrrole N atoms decrease, while those on the pyrrole α-C atom increase as the electron-withdrawing effect of the meso-substituent increases (Table S2). These results can be attributed to a change in the porphyrin orbital of the π-cation radical from the a2u orbital to the a1u orbital with an increase in the electron-withdrawing effect. To evaluate the energy gap between the a1u and a2u radical states, we calculated the energies of these model complexes with three different methods: B3LYP, LC-BLYP, and Hartee–Fock. F0-Cpd I is calculated to the porphyrin π-cation radical in the a2u orbital, the a2u radical state, by all of three methods. However, these three methods afford the different results on the ground spin state having the electron-withdrawing meso-substituent (Fig. 7). The calculations based on the B3LYP show that the a2u radical states are much more stable than the a1u radical states, predicting the a2u ground states for all model complexes. Thus, the switching between the a2u and a1u radical states is not be predicted by the B3LYP. In fact, the previous DFT study on the calculation of 5-Cpd I reported to the a2u radical state, although the present experimental study indicate the a1u radical state [40]. On the other hand, the switching of the ground state is predicted by the Hartree–Fock and LC-BLYP. As the electron-withdrawing effect of the meso-substituent increases, the energy gap between the a2u and a1u radical states decreases and the a2u ground state switches to the a1u ground state at the boundary between 0-Cpd I and 2-Cpd I for the Hartree–Fock and between 2-Cpd I and 3-Cpd I for LC-BLPY. Since the experimental results suggest the switching between 3-Cpd I and 4-Cpd I, the LC-BLPY gives the most precise prediction for the ground state of Cpd I. Although we do not have a correct answer why LC-BLPY is better than B3LYP and Hartree–Fock for the present calculation, an exchange interaction may need to be properly treated in the calculation to obtain a correct energy for Cpd I, because these three functionals have difference in treatment of exchange interaction.

Structures of the iron porphyrin complexes used for DFT calculations of the electron-withdrawing effect of the pyrrole-β substituent

Change of the energies of the Cpd I models having the a1u radical state and the a2u radical state on the electron-withdrawing effect of the meso-substituent. a B3LYP, b Hartree–Fock, c LC-BLYP
We further studied the effect of the electron-withdrawing substituent at the pyrrole β-position. The chemical structures of the model complexes, F4 and F8, are shown in Fig. 6. The optimized structures are also shown in Figure S3. The structural and electronic parameters are summarized in Tables S5–S7. The energies of the ground state and the excited states are summarized in Table S8. The ground states of the Cpd I model complexes of F4 and F8, F4-Cpd I, and F8-Cpd I are the quartet state with an unpaired electron in the a2u orbital. This is confirmed by the Mulliken spin populations of F4 and F8, which are hardly changed by the electron-withdrawing effect (Table S6). In contrast to the electron-withdrawing effect of the meso-substituent, the electron-withdrawing effect of the pyrrole β-substituent does not switch the porphyrin π-cation-radical state (Fig. 8). This is consistent with the previous experimental results [16, 17]. As in the case of the meso-substituent, this has also been attributed to the electron densities at the pyrrole β-position of the a2u and a1u orbitals (Fig. 2) [16, 17, 19].

Change of the energies of the Cpd I models having the a1u radical state and the a2u radical state on the electron-withdrawing effect of the pyrrole-β substituent
Electron-withdrawing effect of porphyrin ligand on the reactivity
To investigate the porphyrin ligand effect on the reactivity, we performed DFT calculations on the chloroiron(III) porphyrin complexes of F0, 2, 3, 5, F4, and F8, which are the final product complexes after the epoxidation reactions of the corresponding Cpd I. The ground spin states of all of the chloroiron(III) porphyrin complexes are the sextet states (ferric high-spin states), and this is unchanged by the electron-withdrawing effect of the meso-substituent. Therefore, the spin state changes from the quartet state to the sextet state during the epoxidation reaction. We calculated the enthalpy changes for the epoxidation reactions of these Cpd Is with ethylene and the calculated values are listed in Tables S9 and S10. Regardless of whether there is a meso-substitution or a pyrrole-β substitution, the enthalpy change of the reaction is greater when the electron-withdrawing effect is stronger. This means that Cpd I becomes more reactive with an increase in the electron-withdrawing effect. To elucidate the mechanism of how the electron-withdrawing substituent activate Cpd I, we compared the change in the enthalpy of formation between Cpd I and the ferric porphyrin complex for F0, F4, and F8. The relative energies for the calculations of the enthalpy changes of formation are listed in Tables S11–S13. These energies indicate how much Cpd I and ferric complex are stabilized by the electron-withdrawing effect of porphyrin ligand. Therefore, these calculations show that both Cpd I and ferric porphyrin complex become more stable as the number of the fluorine atoms at the pyrrole-β position increases. However, the ferric porphyrin complex is stabilized more than Cpd I by the same electron-withdrawing effect, because the difference in the change of the enthalpy of formation between F0 and F8 is 180.9 kJ mol−1 for the ferric porphyrin complex and 162.9 kJmol−1 for Cpd I (Table S12). The difference (18.0 kJ mol−1) between these values results in the difference (18.0 kJ mol−1) in the enthalpy changes of the epoxidation reactions of ethylene by F0 and F8 (Table S13). This means that the electron-withdrawing effect of porphyrin ligand makes Cpd I more reactive by stabilizing the ferric porphyrin more than Cpd I, which produces larger enthalpy change of the reaction of Cpd I.
These DFT calculations suggest that the stability of the sextet state is the key to revealing the porphyrin ligand effect on the reactivity. Therefore, we further calculated the sextet state for Cpd I of F0, F4, and F8. The energies of the sextet state relative to the quartet ground state are summarized in Table S14. The energy gap between the quartet ground state and the sextet state decreases as the electron-withdrawing effect of the pyrrole β-substituent increases (blue arrow in Fig. 9). The change in the energy gap is easily understood by the σ-donor effect of the porphyrin ligand. Since the energy gap corresponds to the excitation energy of the spin from the dxy orbital to the \(d_{{\text{x}^2}-{\text{y}^2}}\) orbital, it is determined by the energy of the \(d_{{\text{x}^2}-{\text{y}^2}}\) orbital. As the electron-withdrawing effect of the substituent increases, the σ-donor effect of the porphyrin ligand becomes weaker and the energy gap between the dxy orbital and the \(d_{{\text{x}^2}-{\text{y}^2}}\) orbital decreases due to the stabilization of the \(d_{{\text{x}^2}-{\text{y}^2}}\) orbital (Fig. 9).

Energy diagram showing the mechanism of how the axial and porphyrin ligands change the reactivity of Cpd I. In this diagram, L, L′, and L″ indicate a series of the axial ligands or porphyrin ligands having different ligand effects. The binding strength of the axial ligand and the electron-withdrawing effect of porphyrin ligand increase in the order of L < L′ < L″
Mechanism for controlling the reactivity of Cpd I
Previously, we have shown that the axial ligand changes the reactivity of Cpd I for oxygenation reactions [39]. The main proposals from the study are as follows (Fig. 9). (1) The binding of the axial ligand alters the stability of both the reactant state (Cpd I) and the product state (the ferric porphyrin), but stabilizes the ferric porphyrin state more than Cpd I state. This is the origin of the free energy of reaction induced by the axial ligand effect. As the binding strength of the axial ligand increases, the free energy of reaction produced by the axial ligand effect is larger. Therefore, Cpd I becomes more reactive as the axial ligand binds to the ferric iron center more strongly. The essence of the axial ligand effect is the stabilization of the ferric porphyrin state. (2) The ground spin state of the ferric porphyrin complex is the key state that controls the reactivity of Cpd I, because the stability of the ground spin state determines the stability of the complex. It is the sextet (ferric high spin) state for most axial ligands, with the exception of thiolate ligands (the ferric low-spin state) and weakly coordinating axial ligands (the ferric intermediate-spin state) [39]. The ground spin state of Cpd I is the quartet state for most axial ligands and thus the ground spin state switches from the quartet state to the sextet state during the oxygenation reactions. As the substrate comes closer to the Fe=O moiety of Cpd I, the Fe=O bond becomes longer and the σ- and π-donor effects of the oxoligand become weaker. This leads to the stabilization of the unoccupied \(d_{\text{z}^2}\) orbital. The sextet state is stabilized, while the quartet state is destabilized with progress in the oxygenation reaction, and finally the ground spin state switches. The switching probably occurs just before the transition state. This proposal has been supported by the experimentally obtained correlation between the free energy of activation and the thermodynamic stability of the ferric porphyrin state, as well as the energy gap between the quartet ground spin state and the sextet-excited state in Cpd I estimated from DFT calculations [39].
The axial ligand of Cpd I also affects the excited energy from the ground quartet state to the excited sextet state. As the axial ligand binds to the ferryl ion stronger, the bond between the porphyrin pyrrole N and the ferryl iron becomes weaker, and the energy of the unoccupied \(d_{{\text{x}^2}-{\text{y}^2}}\) orbital becomes lower. Since the excitation from the quartet state to the sextet state means an electron transition from the dxy orbital to the \(d_{{\text{x}^2}-{\text{y}^2}}\) orbital, the stronger axial ligand makes the excitation energy smaller.
Here, we investigate the effect of the porphyrin ligand on the reactivity of Cpd I by changing the electron-withdrawing effect of the meso-phenyl group. Interestingly, the mechanism of how the porphyrin ligand controls the reactivity of Cpd I is the same as the mechanism of the axial ligand effect. The DFT calculations of ferric porphyrin complexes and Cpd I indicated that electron-withdrawing substituents on the porphyrin ligand stabilize the ferric porphyrin more than Cpd I. This is the origin of the free energy of reaction induced by the porphyrin ligand effect. As the electron-withdrawing effect of the porphyrin ligand increases, the free energy of reaction produced by the porphyrin ligand effect increases. As a result, Cpd I becomes more reactive when the electron-withdrawing effect of the porphyrin ligand is stronger. The switching of the ground spin state from the quartet state to the sextet state also occurs during the oxygenation reaction and the sextet state, the ground spin state of the ferric porphyrin complex, is the key to controlling the reactivity of Cpd I. As in the case of the axial ligand effect, the switching of the ground spin state results from the stabilization of the \(d_{\text{z}^2}\) orbital when transferring the oxygen atom of Cpd I to the olefin in the oxygenation reaction.
Various factors have been proposed to explain the effect of the axial and equatorial ligands on the reactivity of a metal–oxo complex. For example, these are electrophilicity of the oxo ligand, strength or distance of the metal–oxo bond, redox potential of a metal–oxo complex, and the pKa of the oxo ligand. These factors are changed by the ligand effect and consequently alter the activation energy of the reaction by changing the stability of the metal–oxo complex (reactant state). However, these factors are not necessarily correlated with reactivity of compound I. For example, a previous study demonstrate that the axial ligand changes the reactivity of Cpd I without changing the redox potential and Fe=O bond strength [39]. It would depend on the reaction mechanism of the rate-limiting step and the nature of the transition state whether the factor correlates to the reactivity.
References
Poulos TL (2014) Heme enzyme structure and function. Chem Rev 114:3919–3962
Liu W, Groves JT (2015) Manganese catalyzed C-H halogenation. Acc Chem Res 48:1727–1735
Fujii H (2016) Model complexes of heme peroxidases. In: Raven E, Dunford B (eds) Heme peroxidases. The Royal Society of Chemistry, Cambridge, pp 183–217
Puri M, Que L Jr (2015) Toward the synthesis of more reactive S = 2 non-heme oxoiron(IV) complexes. Acc Chem Res 48:2443–2452
Nam W (2015) Synthetic mononuclear nonheme iron-oxygen intermediates. Acc Chem Res 48:2415–2423
Solomon EI, Light KM, Liu LV, Srnec M, Wong SD (2013) Geometric and electronic structure contributions to function in non-heme iron enzymes. Acc Chem Res 46:2725–2739
Groves JT, Haushalter RC, Nakamura M, Nemo TE, Evans BJ (1981) High-valent iron-porphyrin complexes related to peroxidase and cytochrome P-450. J Am Chem Soc 103:2884–2886
Balch AL, Latos-Grazynski L, Renner MW (1985) Oxidation of red ferryl [(FeIVO)2+] porphyrin complexes to green ferryl [(FeIVO)2+] porphyrin radical complexes. J Am Chem Soc 107:2983–2985
Sugimoto H, Tung H, Sawyer DH (1988) The formation, characterization, and reactivity of the oxene adduct of [tetrakis(2,6-dichlorophenyl)porphinato]iron(III) perchlorate in acetonitrile. Model for the reactive intermediate of cytochrome P-450. J Am Chem Soc 110:2465–2470
Gross Z, Nimri SA (1994) A pronounced axial ligand effect on the reactivity of oxoiron(IV) porphyrin cation radicals. Inorg Chem 33:1731–1732
Gross Z, Nimri S, Barzilay CM, Simkhovich L (1997) Reaction profile of the last step in cytochrome P-450 catalysis revealed by studies of model complexes. J Biol Inorg Chem 2:492–506
Gold A, Jayaraj K, Doppelt P, Weiss R, Chottard G, Bill E, Ding X, Trautwein AX (1988) Oxoferryl complexes of the halogenated (porphinato)iron catalyst [tetrakis(2,6-dichlorophenyl)porphinato]iron. J Am Chem Soc 110:5756–5761
Mandon D, Weiss R, Jayaraj K, Gold A, Terner J, Bill E, Trautwein AX (1992) Models for peroxidase compound I: generation and spectroscopic characterization of new oxoferryl porphyrin π cation radical species. Inorg Chem 31:4404–4409
Fujii H (1994) Characterization of high valent iron porphyrin in catalytic reaction by iron(III) tetrapentafluorophenylporphyrin. Chem Lett 23:1491–1494
Cong Z, Kurahashi T, Fujii H (2011) Oxidation of chloride and subsequent chlorination of organic compounds by oxoiron(IV) porphyrin π-cation radicals. Angew Chem Int Ed 50:9935–9939
Fujii H (1993) Effects of the electron-withdrawing power of substituents on the electronic structure and reactivity in oxoiron(IV) porphyrin π-cation radical complexes. J Am Chem Soc 115:4641–4648
Fujii H, Ichikawa K (1992) Preparation and characterization of an A1u (Oxo) iron(IV) porphyrin π-cation-radical complex. Inorg Chem 31:1110–1112
Takahashi A, Kurahashi T, Fujii H (2009) Effect of imidazole and phenolate axial ligands on the electronic structure and reactivity of oxoiron(IV) porphyrin π-cation radical complexes: drastic increase in oxo-transfer and hydrogen abstraction reactivities. Inorg Chem 48:2614–2625
Fujii H, Yoshimura T, Kamada H (1997) Imidazole and p-nitrophenolate complexes of oxoiron(IV) porphyrin π-cation radicals as models for compounds I of peroxidase and catalase. Inorg Chem 36:6142–6143
Traylor TG, Xu F (1988) Catalytic asymmetric epoxidations with chiral iron porphyrins. J Am Chem Soc 110:1953–1958
Traylor TG, Nakano T, Dunlap BE, Traylor PS, Dolphin D (1986) Mechanisms of hemin-catalyzed alkene epoxidation. The effect of catalyst on the regiochemistry of epoxidation. J Am Chem Soc 108:2782–2784
Traylor TG, Nakano T, Mikzstal AR, Dunlap BE (1987) Transient formation of N-alkylhemins during hemin-catalyzed epoxidation of norbornene. Evidence concerning the mechanism of epoxidation. J Am Chem Soc 109:3625–3632
Collman JP, Brauman JI, Meunier B, Rayback SA, Kodadek T (1984) Epoxidation of olefins by cytochrome P-450 model compounds: mechanism ofoxygen atom transfer. Proc Natl Acad Sci USA 81:3245–3248
Collman JP, Brauman JI, Meunier B, Hayashi T, Kodadek T, Rayback SA (1985) Epoxidation of olefins by cytochrome P-450 model compounds: kinetics and stereochemistry of oxygen atom transfer and origin of shape selectivity. J Am Chem Soc 107:2000–2005
Collman JP, Kodadek T, Rayback SA, Brauman JI, Papazian LM (1985) Mechanism of oxygen atom transfer from high valent iron porphyrins to olefins: implications to the biological epoxidation of olefins by cytochrome P-450. J Am Chem Soc 107:4343–4345
Fujii H, Yoshimura T, Kamada H (1996) ESR studies of A1u and a 2u oxoiron(IV) porphyrin π-cation radical complexes. Spin coupling between ferryl iron and A1u/a 2u orbitals. Inorg Chem 35:2373–2377
Kitagawa T, Mizutani Y (1994) Resonance Raman spectra of highly oxidized metalloporphyrins and heme proteins. Coord Chem Rev 135–136:685–735
Czarnecki K, Proniewicz LM, Fujii H, Kincaid JR (1996) Resonance Raman spectrum of a 2A1u ferryl porphyrin π-cation radical. J Am Chem Soc 118:4680–4685
Czarnecki K, Minri S, Gross Z, Proniewicz LM, Kincaid JR (1996) Direct resonance Raman evidence for a trans influence on the ferryl fragment in models of compound I intermediates of heme enzymes. J Am Chem Soc 118:2929–2935
Czarnecki K, Fujii H, Kincaid JR (1999) Resonance Raman spectra of legitimate models for the ubiquitous compound I intermediates of oxidized heme enzymes. J Am Chem Soc 121:7953–7954
Boso B, Lagn G, McMurry TJ, Groves JT (1983) Mössbauer effect study of tight spin coupling in oxidized chloro-5,10,15,20,-tetra(mesityl)porphyrinateiron(III). J. Chem. Phys. 79:1122–1126
Jones RJ, Jayaraj K, Gold A, Kirk ML (1998) Ground and excited state spectroscopic probes of [(TMP)Fe(IV) = O] + : the first magnetic circular dichroism study of a model peroxidase compound I intermediate. Inorg Chem 37:2842–2843
Fujii H (2002) Electronic structure and reactivity of high-valent oxo iron porphyrins. Coord Chem Rev 226:51–60
Asaka M, Fujii H (2016) Participation of electron transfer process in rate-limiting step of aromatic hydroxylation reactions by compound I models of heme enzymes. J Am Chem Soc 138:8048–8051
Lindsey JS, Wagner RW (1989) Investigation of the synthesis of ortho-substituted tetraphenylporphyrins. J Org Chem 54:828–836
Wylie GRA, Munro OQ, Schulz CE, Scheidt WR (2007) Structure and physical characterization of (nitrato)iron(III) porphyrinates [Fe(por)(NO3)]–variable coordination of nitrate. Polyhedron 26:4664–4672
Gaussian 09, Revision E.01, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian, Inc., Wallingford
Patterson WR, Poulos TL, Goodin DB (1995) Identification of a porphyrin π cation radical in ascorbate peroxidase compound I. Biochemistry 34:4342–4345
Takahashi A, Yamaki D, Ikemura K, Kurahashi T, Ogura T, Hada M, Fujii H (2015) Effect of the axial ligand on the reactivity of the oxoiron(IV) porphyrin π-cation radical complex: higher stabilization of the product state relative to the reactant state. Inorg Chem 51:7296–7305
Sinna MA, Kumar S, Kumar D, Fornarini S, Crestoni ME, de Visser SP (2015) A comprehensive test set of epoxidation rate constants for iron(IV)–oxo porphyrin cation radical complexes. Chem Sci 6:1516–1529
Acknowledgements
This study was supported by grants from JSPS (Grant nos. 25288032 and 17H03032) and CREST. We thank IMS for assistance of NMR and EPR measurements.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ishimizu, Y., Ma, Z., Hada, M. et al. Experimental and theoretical studies of the porphyrin ligand effect on the electronic structure and reactivity of oxoiron(iv) porphyrin π-cation-radical complexes. J Biol Inorg Chem 24, 483–494 (2019). https://doi.org/10.1007/s00775-019-01664-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-019-01664-3