
Abstract
In this paper, development and optimization of a new method for determination of the three hormones (estrone, 17-β-estradiol and 17-α-ethinylestradiol) of the Watch list were described. The validated method relies on isotope dilution—two-step solid phase extraction–liquid phase chromatography mass spectrometry method. The measurement procedure validation has been performed in real matrices, including matrix with high suspended particulate matter and high organic carbon contents, to demonstrate its fitness for purpose. Method performances were in agreement with the requirements of the decision 2015/495/EU (maximal acceptable method detection limit and whole-water analysis). Limits of quantification of the method were of 0.4 ng L−1 for estrone and 17-β-estradiol and 0.1 ng L−1 for 17-α-ethinylestradiol. Expanded uncertainties (k = 2) at limit of quantification were equal to 35 % for estrone and 17-β-estradiol and 50 % for 17-α-ethinylestradiol. The method was successfully implemented to monitor French inland surface waters contamination. The survey reveals a chronic state of contamination by estrone (96 % quantification rate) and significant one by 17-β-estradiol and 17-α-ethinylestradiol (15 % quantification rate). Maximum measured concentrations are above predicted no-effect concentrations, indicating potential risk toward the environment.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
According to the Directive 2013/39/EU [1], a new mechanism is needed to provide high-quality monitoring information on the concentrations of potentially polluting substances in the aquatic environment to support future prioritization exercises and thereby to improve the protection of the aquatic environment and of human health via the environment. The mechanism is aimed at emerging pollutants and other substances for which the available monitoring data are either of insufficient or of insufficient quality for the purpose of identifying the risk posed across the European Union (EU). It involves creating a Watch List with a limited number of such substances and monitoring them EU-wide for up to 4 years. A maximum number of ten substances or groups of substances shall be included in the first Watch list, increasing by one at each update, up to a maximum of 14 substances or groups of substances. Frequent reviews of the list will ensure that substances are not monitored longer than necessary, and that substances for which a significant risk at EU level is confirmed are identified as candidate priority substances with as little delay as possible. Three hormones such as 17-β-estradiol (E2), 17-α-ethinylestradiol (EE2) together with estrone (E1) have been selected for inclusion in this first list for the purpose of facilitating the implementation of appropriate measures to address the risk posed by those substances [2].
E2 is the predominant natural female sex hormone and is the most active of the naturally occurring estrogenic hormones and is also a key intermediate in industrial synthesis of other estrogens and of various hormonal 19-norsteroids. E1 is a natural female sex hormone and also an oxidation product of E2. EE2 is a synthetic hormone derived of the natural estrogen E2 which is mainly used as a component of oral contraceptives. Other applications of EE2 in human medicine include estrogen replacement therapy and suspension of breast feeding [3, 4]. Indeed, the presence of E2 and EE2 in the environment has been associated with fish feminization (e.g., synthesis and secretion of vitellogenin), reproduction and behavior modifications, fertility reduction, increase of breast and testicular cancer in humans and promotion of abnormal reproductive processes [5].
Physicochemical properties of substances especially solubility in water (Sw) and the octanol/water partition coefficient (Kow) are considered crucial to assess the fate of chemicals in the environment. In general, chemicals with Kow values < 10 are considered relatively hydrophilic, tending to have high Sw and low adsorption and bioconcentration factors. On the opposite, compounds with Kow values > 104 are considered very hydrophobic and have high sorption potential, such as E2 and EE2 with log Kow values around four (Table 1). Accordingly, the 3 substances have the potential to disseminate within aquatic environments, to enter the food chain and to bioaccumulate [5].
The monitoring of the substances in the Watch list should generate high-quality data on their concentrations in the aquatic environment, fit for the purpose of supporting the risk assessments that underpin the identification of priority substances. The method detection limit should be at least as low as the substance-specific predicted no-effect concentration (PNEC) for each substance in the relevant matrix. For comparability, all substances should be monitored in whole-water samples (including colloidal fractions and suspended particulate matter) [2].
Loos [4] highlighted that the lowest limit of quantification (LOQ) reported in the literature both for E2 and EE2 is 0.05 ng L−1, which is enough to reach the annual average environmental quality standard (AA-EQS) of E2 (0.4 ng L−1) in inland surface waters, but not sufficient for EE2 (AA-EQS 0.035 ng L−1) (Table 1). For E1, the PNEC of 3.6 ng L−1 is easily achievable by conventional techniques. The achievement of maximum acceptable method detection limit (MDL) in the low pg L−1 concentration range is extremely difficult, if not impossible with current analytical methods (Table 1).
To meet the requirements of comparable and high-quality measurements, the following issues have to be cautiously scrutinized.
Stability
Chemical monitoring relies on a succession of actions: sampling, storage and preservation of representative samples; pre-treatment of a sample portion for quantification, calibration, final determination, calculation of results, uncertainty estimation and final expression of results. Throughout this chain of operations, the guarantee of the stability of the analyte is a key issue that has to be addressed to correctly qualify and discuss monitoring data.
As highlighted by Mompelat et al. [6], from one study to another, controversial results are observed. EE2 is still resistant to sorption and degradation and accordingly its stability in unpreserved samples and uncontrolled temperature conditions can be assumed, but this is obviously not the case for E2 and E1. In fact, for both substances, significant differences in the data suggest that the individual composition of tested water samples (microbial flora, pH, organic matter, minerals, etc.) within study (lack of homogeneity of the material) and between different studies may vary and thus affects kinetics of degradation. Various sample pre-treatments (filtration, freezing) and preservation techniques with both acidic and non-acidic agents have been tested with variable degrees of success [6]. Meanwhile, most of these strategies were thought with a research point of view and not with an operational routine one (when sampling operations are handled by accredited samplers). Such considerations represent a critical point in the context of the present work.
The whole-water analysis
It appears necessary to remind that the requirements are to monitor substances in the whole-water, that is to say including suspended particulate matter (SPM). Nie et al. [7] determined the intrinsic association of estrogen compounds among different water phases: suspended particle matter (> 1 μm), filtrate (< 1 μm), colloidal (between 1 KDa and 1 μm) and soluble phases (< 1 kDa) using a validated cross-flow ultra filtration system. Their results highlighted that, depending on the type of matrix and season, 5 % to 75 % of E1, 5 % to 35 % of E2 and up to 100 % of EE2 were associated with SPM fractions. Moreover, they also showed that 2.0 % to 58.4 % of E1, 8.36 % to 72.0 % of E2 and up to 20.6 % of EE2, were associated with colloidal fractions, and that organic carbon content strongly affects the partition of estrogens in water samples. These observations confirmed that colloids and SPM could act as a significant sink for environmental estrogens and may participate to their transport in/through water systems. In 2015, the same authors carried out a study on the occurrence, distribution and risk assessment of six estrogen substances in samples of surface water, SPM, and sediment in the Yangtze Estuary and its coastal areas over four seasons. The SPM phase for total estrogens contributed between 16 and 66 % in July, 24 % to 63 % in October, 21 % to 54 % in January, and 18 % to 88 % in May. Moreover, based on a yeast estrogen screen (YES) bioassay, SPM and sediment phase were shown to exhibit higher estrogenic risks compared to the water phase [8]. Sangster et al. [9] suggested that particle interactions might cause a preferential distribution of hormones within fine particle size fractions of whole sediments especially when they are present at low concentration level (1 ng L−1). As a matter of fact, estrogens associated with these particles are more likely to travel greater distances. The authors concluded that aquatic organisms may have more contact, through ingestion or respiration across gills, with smaller particles, thus, increasing their exposure to hormones. Yang et al. [10] studied the partitioning behaviors of endocrine disrupting chemicals (EDC), among while estrogenic substances, between the solid phase [sediment, fine SPM (≥ 0.7 μm < 2.7 μm), coarse SPM (≥ 2.7 μm)] and the dissolved phase (< 0.7 μm). Their ambition was to elucidate the relationships between concentrations and partitions of EDC in different matrix. Compared to the dissolved phase, partition of most EDC in the solid phase was influenced by particle size. Concentrations of EDC were negatively correlated to the amount of particles in the respective fraction, which is so-called “particle concentration” effect. In a review on methodological challenges of multi-residue analysis of pharmaceuticals in environmental samples, Petrovic [11] pointed out the main drawbacks and challenges on current trends in environmental analytical chemistry; among them, the issue of whole-water analysis as required by Water Framework Directive (WFD). This issue has also been discussed by Ademollo et al. [12]. The authors recommended that total concentration should be obtained by direct analysis of the whole-water sample (without filtration) or by a separate determinations on filterable (dissolved) and solid phases (particulate). They highlighted filtration step might introduce significant loss of compound due to sorption of analytes on the filter material, especially for estrogens. Similarly, Neale et al. [13] showed that no less than 50 % of E2 could be lost by filtration through cellulose acetate filters. De Graaff et al. [14] reported up to 64 % adsorption of estrogens when filtrated over a glass fiber filter (E1, E2, EE2).
In a review of Loos [4], the most popular approach to analyze estrogens in environmental aqueous matrix is solid phase extraction SPE cartridges on line or off-line, with or without a filtration step. Accordingly, the question about the relevance of these methodologies in the context of WFD monitoring requirements can be pointed out, especially regarding its capability to take into account SPM.
Solid phase extraction (SPE) disks are used in many application fields as modified versions of the widespread SPE cartridges. The sorbent particles embedded in the disks are much smaller than those used for filling the cartridges enabling increased extraction efficiency as well as minimized SPE drawbacks (limited flow rate of the sample, risks of loss of analytes, important void volume; risks of channeling). Moreover; SPE disks offer better capability to deal with SPM compared with SPE cartridges that are well known to suffer from plugging. SPE disks look promising [15] in particular to face current trends in legislation and normalization in the context of implementation of the WFD. As proof, three European standards have been recently published implementing SPE-disk-based extraction techniques and demonstrating their fitness for purpose to monitor WFD priority substances [16,17,18]. Moreover, it has been successfully implemented for estrogenic compounds monitoring [19, 20].
Sensitivity
As has been emphasized previously, these three estrogenic substances have to be analyzed at sub ng L−1 concentration level; but the lack of available procedures has been obviously highlighted and is consensually admitted. Accordingly, it appeared necessary to build a new analytical strategy to overpass the limitations, keeping in mind the needs of WFD implementation. Qi et al. [21] reviewed the issue of chemical derivatization-based liquid chromatography–mass spectrometry (LC–MS) and highlighted that it is a promising strategy to solve many analytical problems, especially the lack of sensitivity in consequence of poor ionization efficiency. Estrogens are phenolic compounds that may suffer from poor ionization in electrospray ionization. Accordingly, to improve the MS signal, derivatization has been widely used to increase ionization efficiency but not for environmental monitoring purpose, contrary to human or food regulatory monitoring [22]. Lien et al. [23] investigated signal intensity and matrix effects (different natural waters: effluents, surface water) on various chromatographic systems and several ionization modes for both estrogenic compounds and their derivatives (dansyl chloride and pentafluorobenzyl bromide). They pointed out that dansyl derivatization significantly enhances specificity and sensitivity of analysis and appears promising for estrogens ultratraces monitoring, in accordance with Zhang et al. [24].
The present work aims at presenting method optimization and validation to enable a reliable quantification of three targeted estrogenic compounds (E1, E2 and EE2) according to Decision (EU) 2015/495 [2]. Relevant steps of the method are described in this work as well as comprehensive development to insure data reliability. Finally, the validated method was implemented for the measurement of targeted compounds in French inland surface waters in the context of Watch list monitoring activity. Preliminary results on the occurrence of the targeted estrogens are presented.
Materials and methods
Chemicals and reagents
The set of native and labeled analytical standards (C13 or Deuterated) was purchased as pure standards from Sigma–Aldrich (Saint-Quentin Fallavier, France) (see SI Table S1). Doing so, isotope dilution that is recognized to be the most efficient way to compensate all matrix effects since they affect both and indistinctly the analyte and its labeled analog, was implemented.
Acetonitrile (Ultra gradient HPLC—grade Baker HPLC analyzed) and methanol (MeOH) (Baker analyzed LC–MS reagent), acetone (AnalaR Normapur®) were acquired from Atlantic labo-Ics (Bruges, France). High-purity laboratory water was provided by a Milli-Q purification system (Millipore, Molsheim, France). Evian® table water was used as “reference” water for spiked samples during the method development and validation. Formic acid (AnalaR Normapur®, purity > 99 %), hydrochloric acid 37 % (Merck), Dansyl chloride (for HPLC derivatization, 99 %) from VWR (Fontenay-sous-Bois, France) and from Sigma–Aldrich (Saint-Quentin Fallavier, France).
For method validation, two types of water were used: Evian® table water as low complexity matrix and Oise River (SPM > 100 mg L−1; dissolved organic carbon > 5 mg L−1) as high complexity water.
Stock solutions preparation
All stock solutions were prepared gravimetrically following the procedures of the French National Institute of Metrology (LNE). Each weighing was repeated five times for masses up to 5 mg and three times for higher masses. Individual stock solutions of each drug residue and its labeled analog were prepared at about 100 μg mL−1 in methanol. Mixed solutions of the three native surrogates and of the three isotopically labeled surrogates were then prepared in methanol at between 50 and 100 ng mL−1, respectively. All stock solutions were stored at (− 20 ± 6) °C in the dark. Their stability was checked and guaranteed.
Sampling, storage and transport
Sampling operations were carried out by accredited samplers in parallel to current WFD surveillance monitoring sampling campaigns. Samplers follow French national guidance on sampling. Samples were kept in amber glass bottles, previously calcinated at 450 °C, 3 h. They were stored and transported at 4 ± 3 °C. Samples were taken in hand for sample preparation within 12 to 24 h post-sampling.
Sample preparation: extraction/purification/derivatization
Before extraction, 50 μL of the mixed solution of labeled compounds was added volumetrically to each sample. Solid phase extraction was achieved on Bakerbond H2O-Philic DVB Speedisk® (Interchim, France) on a Horizon Technology SPE-DEX 4790 Automated Extraction System SPE-DEX® 4790 according to the program in SI Table S2. Eluates were then evaporated to 1 mL under vacuum at a temperature of 40 °C using a Savant SC250Exp SpeedVac® concentrator (Thermo-Scientific, Villebon-sur-Yvette, France). Methanolic extracts were then purified by solid phase extraction on aminopropyl (LC-NH2 SUPELCO 500 mg, 6 cc). Each cartridge was rinsed with 4 mL methanol, and then, the 1 mL extract was loaded and kept on a receiving vial. Cartridge was washed with 2 mL of methanol. Eluates were then evaporated to dryness under vacuum at a temperature of 40 °C using a Savant SC250Exp SpeedVac® concentrator before being derivatized.
The extracts were taken up in 200 μL of acetone and 500 μL of sodium bicarbonate buffer (100 mM, pH = 10.5) was added. The mixture was stirred by vortex for around 1 min. A volume of 500 μL of dansyl chloride solution (1 mg mL−1 in acetone) was added. The mixture was stirred by vortex for one min before being heated to 60 °C for 6 min and evaporated to dryness with SpeedVac® (kinetics and efficiency of derivatization were studied and verified, data not shown). The extracts were taken up in 2*600 μL MeOH in a new vial in order to remove insoluble salts before being evaporated to dryness, then taken by 100 μL of mixture of water-acetonitrile (35/65; v/v).
LC–MS/MS analysis
Liquid chromatography analysis was performed on a Surveyor LC/TSQ Quantum Discovery max (Thermo Fischer Scientific). More specifically, separation was achieved on C18 Symmetry Shield® column (RP18, Waters, 3.5 μm, 150 mm×2.1 mm) heated at 20 °C. Solvent A was acidified milli-Q water (0.1 % (v/v) formic acid), and solvent B was acetonitrile with 0.1 % (v/v) formic acid. The mobile phase flow was set to 0.4 mL min−1. Separation was achieved implementing a 20 min isocratic gradient 35 % A/65 % B.
Mass spectrometry analysis was achieved by a triple quadruple mass spectrometer equipped with an electrospray ionization (ESI) source operated in positive mode. The mass spectrometer optimized parameters are detailed in SI Table S3. Acquisition was performed in MRM mode. Tandem mass spectral analysis of the MH+ ion of dansyl-estrogen provided a major product ion (m/z 171) arising from fragmentation of the dansyl group. Protonated molecular ion of the derivatized form was chosen as precursor, whereas first and second most abundant ion transitions were selected for quantification and confirmation, respectively. Selectivity aside, the dansyl derivatization allows detection and quantification of sub-part per trillion levels of estrogens in environmental water matrix. It must be suitable for an accurate and reliable quantification in various complexity matrixes if an efficient extraction and purification step is implemented.
Method characterization
In France, NF T90-210 standard [25] for water quality is mandatory to operate in water monitoring. It defines a “protocol for the initial method performance assessment in a laboratory”. In other words, it provides laboratories specific statistical tools for the initial evaluation of either developed, adapted or standardized quantitative methods in the field of water physicochemical analysis [25]. Accordingly, method validation was performed, following this standard, for relevant performances for its application: recovery, limit of quantification, accuracy. To allow the comparison of measurements, it is necessary to display of a realistic uncertainty estimation of the measurements. In fact, uncertainty expresses the doubt, inherent to any measurement process, on a given measurement as X ± U (k = y) where U is the expanded measurement uncertainty and y the coverage factor. Typically, a coverage factor of 2 is applied ensuring a confidence interval of 95 %. Accordingly, measurement uncertainties of the method were evaluated taking into account the main sources of uncertainty, according to the ISO 11352 [26].
Calibration model analysis
Multipoint calibration curves were gravimetrically prepared in a mixture of Milli-Q water 0.1 % (v/v) formic acid and acetonitrile 0.1% (v/v) formic acid (65/35; v/v). Analyte concentrations ranged from 0.4 ng mL−1 and 11 ng mL−1 for EE2, 1 ng mL−1 and 36 ng mL−1 for E1 and E2. Each point was randomly injected in duplicate. Calibration curves have been determined in intermediate precision conditions during six different days. Calibration models were then analyzed according to NF T90-210 standard [25].
Limits of quantification (LOQ)
The standard NF T90-210 [25] defines that it is necessary to check that presupposed LOQ is acceptable, with respect to a maximal admissible deviation of 60 %, in natural representative matrix. This check must imperatively be performed in the conditions of application of the method. Accordingly, the estimated quantification limits were verified by analyzing both reference laboratory waters (Evian® Water) and representative environmental samples (surface water from the Oise River) spiked at pre-estimated limit of quantification for each compound, in intermediate precision conditions (two operators, six different days, two replicates).
The accuracy of the presupposed LOQ is verified regarding a maximum acceptable deviation of 60 % by checking the following two inequalities (1 and 2) [25]:
where ZLOQ is the average calculated concentration, SLOQ is the calculated standard deviation in intermediate precision conditions.
If at least one of the inequalities is not checked, then the LOQ is not verified.
Method recoveries
Recoveries were determined by analyzing both reference laboratory waters (Evian® table water) and representative environmental samples (surface water from the Oise River). The tests were carried out independently by spiking samples at MDL, 3MDL and 6MDL (equivalent to 0.4 ng L−1, 1.2 ng L−1, 3.6 ng L−1for E1 and E2; 0.035 ng L−1, 0.1 ng L−1, 0.4 ng L−1 for EE2) in intermediate precision conditions (two operators, 6 days, duplicate analysis).
Method accuracy
The measurement accuracy is defined as the “closeness of agreement between a measured quantity value and a true quantity value of a measurand”. The concept of accuracy is related to both measurement trueness and precision. In other words, to be considered accurate, a method should provide measured results characterized for their “closeness of agreement” to “the reference quantity value” (trueness) and to “quantity values obtained by replicate measurements” (precision). Without any available matrix-based Certified Reference Material, the purchased analytical standards were used as reference. Independent replicate of Evian® water sample (1000 mL) and Oise River (1000 mL) spiked at MDL, 3MDL and 6MDL were analyzed during six different days. Maximum percentage tolerance to validate accuracy of the method was fixed depending on the compound and the level of concentration.
Uncertainties evaluation
To allow the comparison of measurements, it is necessary to display of a realistic uncertainty estimate of the measurements. Accordingly, measurement uncertainties of the method were evaluated taking into account the main sources of uncertainty, according to ISO 11352 [26], implementing MUKIT software [33].
Results and discussion
During method optimization, each step of the overall method has been rigorously addressed in order to ensure of its effectiveness and reliability as necessary to display of a robust analytical method.
Stability of compounds
Stability of the three targeted analytes in matrix was checked in order to validate the national strategy. Two representative matrices (Evian water, Seine River water) were selected and two storage conditions were tested (freezing (− 20 ± 6) °C for midterm storage, and 4 ± 3 °C for 24 h). Samples were spiked at 3 MDL. For each tested conditions, triplicate samples were analyzed. All sampled were analyzed randomly under repeatable conditions (same instrumental run). Depending on the compound and the condition, average losses were comprised between 7 and 26 % in 24 h (data not shown).These preliminary results have helped to design the following sample management strategy. Samples were transported, at 4 ± 3 °C, to laboratory in less than 24 h following sampling. At arrival, 1 L of sample was immediately engaged in the analytical procedure and 1 L was stored at (− 20 ± 6) °C for confirmation of results, if necessary.
Overall method recovery
Overall method recoveries were determined by analyzing a synthetic matrix (Evian®) and natural matrix (Oise River with a high level TSS content around 100 mg L−1). The tests were carried out independently by spiking samples at 3 different levels of concentrations (MDL, 3MDL and 6MDL) in intermediate precision conditions (two operators, 6 days) (Fig. 1). For E1 and E2, whatever the level of concentrations and the matrix, recoveries were comprised between 79 and 120 % with CV around 10 % in intermediate precision conditions. For EE2, the picture is different and illustrates the difficulty to obtain accurate and reliable measurements at ultratraces level. At 3 MDL and 6 MDL, recoveries of the method were comprised between 67 and 127 % and CV below 25 %. On the contrary, at MDL (0.035 ng L−1), recoveries of the method can reach 200 % with high CV. These observations can easily be justified by the fact that targeted concentrations between detection limit and quantification limit of the method (see discussion in paragraph 3.3) and the measurement are significantly affected by the background signal noise of the samples. Isotope dilution demonstrated its relevance to achieve high-precision analyses. For the three targeted substances, no operator effects neither day effects were observed. It is always difficult to compare method performances considering the diversity of target compounds, terminologies, and methodologies of determination found in the literature. Method performances, high extraction rates with low precision, were considered as suitable to meet the objectives of method implementation. In this regard, the results presented here are satisfactory in terms of the state of the art in the literature. Other methods developed for the quantification of some of the investigated compounds in waters reported comparable method recoveries [4, 20].

The figure presents the recovery of each tested conditions [days, operators (op), replicates (rep), and level of concentrations (MDL, 3 MDL and 6 MDL see paragraph 2.6.3 for more information)] for the different tested water matrices. The white bars on the right part of the figure presents the mean recovery in intermediate conditions precision in Evian® table water
Limit of quantification
Operational LOQs of the method were determined and are summarized in Fig. 2 and Table 2. They were considered as satisfactory based on the objectives of the method: 0.4 ng L−1 for E1 and E2 and 0.1 ng L−1 for EE2. The comparison of method performances, especially the LOQ, is critical as many different vocabularies and estimation strategies are found in the literature [27]. With a S/N = 10 approach to estimate LOQs in mineral water, Devier et al. [28] reported a LOQ of 1 ng L−1 for estrogens. With a similar approach, Esteban et al. [29] reported LOQ 0.12 ng L−1 for E2, 0.17 ng L−1 for E1 and 0.47 ng L−1 for EE2. Avar et al. [30] determined LOQ (signal to noise = 10) from standard solutions and found values equal to 0.05 ng L−1 (MS1) and 0.1 ng L−1 (MS2) by E2, and 0.001 ng L−1 (MS1) and 0.2 ng L−1 (MS2) by EE2. Tavazzi et al. [31] obtained a LOQ for EE2 in Milli-Q water (0.03 ng L−1) that meets the WFD criteria. However, when implementing their method in surface water, the LOQ for EE2 reached 0.07 ng L−1 which is the lowest reported LOQ for determination of EE2 in aqueous samples. Česen and Heath [20] developed a large volume extraction (10L)-SPE disk-GC–MS method, they reported LOQ of 0.290 ng L−1 for E1, 1.37 ng L−1 for E2, 0.724 ng L−1 for EE2. Despite extracting 10 L of sample, LOQ for E2 and EE2 did not achieve the requirements.

The figure displays for each targeted compound, the accuracy profile at three different levels of concentrations including method detection limit (MDL) and limit of quantification (LOQ). EMA: Maximum acceptable error. At LOQ, the EMA has been fixed at 60 % as defined in NF T90-210 standard, at upper level of concentration EMA has been fixed with respect to the 50 % maximum uncertainty
Method accuracy and uncertainty of measurements
As shown in Fig. 2, considering results in terms of trueness and precision with a 30 % tolerance, accuracy of the method was demonstrated for E1 and E2 at concentrations levels of 3MDL and 6 MDL. For EE2, accuracy of the method was demonstrated considering a 40 % tolerance at concentrations level of 6MDL.
Expanded measurement uncertainties (k = 2), at the targeted LOQ, ranged between 50 % for EE2 and 30 % for E1 and E2. Considering the targeted level of concentrations, they were considered as satisfactory. To the authors’ knowledge, there is no published work to which uncertainties can be compared with regarding the investigated compounds in comparable conditions. A reference method developed by the United Kingdom National Institute of Metrology (LGC) aiming at the quantitative determination of five synthetic estrogens in surface waters showed measurement uncertainties between 4 and 12.5 % (unpublished data), but the level of concentrations and measurement strategy is not strictly comparable.
The procedure based on an Isotope Dilution—two-step SPE-LC–MS/MS method allowed for quantification of the three target estrogens in surface waters. The method showed performances in terms of LOQ, uncertainty and matrix that were considered sufficient for its intended uses. Accordingly, it was implemented in the French Watch list monitoring survey.
Application in a national survey
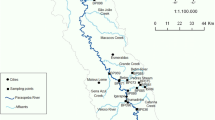
In France, 26 stations were selected for the Watch List monitoring (Fig. 3). They were selected by French water agencies for their representativeness for contrasting pressures (industrial, urban and agricultural). Sampling campaign took place in spring 2016. Table 3 presents an overview of the quantification frequency, min, max, mean and median concentrations. It reinforces the criticality of displaying of methods suitable to the needs and stakes of monitoring in order to answer appropriately to the risk posed by the occurrence of these molecules. This statement is supported when looking at median concentrations that are higher than or closed to EQS or PNEC values (see Table 1).

The figure shows the location of the 26 investigates sites all over the French territory
A screening study on emerging contaminants was carried out in 2012 in surface waters in both metropolitan France and overseas departments. Low quantification frequency (5.6 % for E1 and 0.6 % E2) were observed, but LOQ of the study (1 ng L−1 E2 and 5 ng L−1 for E1) were higher than in this one. Median concentrations of 3 ng L−1 for E1 and 1 ng L−1 for E2 were estimated (Botta, study available on French Biodiversity Agency’ website). In the exploratory campaign organized by the JRC (more than 100 rivers in 27 countries) [32], estrone was quantified in 16 % of the samples at an average concentration of 4 ng L−1 (maximum value = 81 ng L−1, percentile 90 = 10 ng L−1). E2 was also detected in all samples but at lower concentrations (≤ 2 ng L−1). The synthetic hormone, EE2, was detected only at the output of wastewater treatment plant, but its concentration was not quantifiable (LOQ = 1.2 ng L−1). In the context of Watch list, Česen and Heath [20] did not quantify any of the three estrogenic compounds in Slovenian surface water. It has to be pointed out that for E2 and EE2, the developed method did not achieve the requirement of [2].
Conclusions
An isotope dilution—two-step solid phase extraction–liquid phase chromatography mass spectrometry method, together with stringent quality control and quality assurance protocols, was developed and validated according to French mandatory standards in real matrix. Method performances were in agreement with the requirements of the decision 2015/495/EU [2]. LOQ of the method were of 0.4 ng L−1 for E1 and E2 and 0.1 ng L−1 for EE2. Expanded uncertainties (k = 2) at LOQ were equal to 35 % for E1 and E2 and 50 % for EE2. Although validated with respect to French mandatory requirement, the validated method can also be implemented in other European countries or worldwide. The study highlights the complexity of reaching confident data, through reliable and accurate measurements, at the ultratrace level in a real matrix. The developed method was successfully implemented to monitor French inland surface waters contamination by E1, E2 and EE2. The survey reveals a chronic state of contamination by E1 and significant one by E2 and EE2. The measured environmental concentrations legitimate the relevance of the three substances for environmental monitoring considering the potential risk links to their occurrence in aquatic system. It justifies the need to pursue monitoring actions to better understand their fate and occurrence in water systems. Moreover, some additional works are needed to harmonized and standardized analytical methods to sustain the need of comparability of measurements within EU Member states.
References
European Union (2013) Directive 2013/39/EU of the European Parliament and the Council of 12. August 2013 Amending Directives 2000/60/EC and 2008/105/EC as Regards Priority Substances in the Field of water policy. OJ L226, p 1, Accessed 24 Aug 2013
European Union (2015) Decision (EU) 2015/495 of 20 March 2015 establishing a watch list of substances for Union-wide monitoring in the field of water policy pursuant to Directive 2008/105/EC of the European Parliament and of the Council
Barreiros L, Queiroz JF, Magalhães LM, Silva AMT, Segundo MA (2016) Analysis of 17-β-estradiol and 17-α-ethinylestradiol in biological and environmental matrices—a review. Microchem J 126:243–262
Loos R (2015) Analytical methods for possible WFD 1st watch list substances. EUR 27046 EN
Adeel M, Song X, Wang Y, Francis D, Yang Y (2017) Environmental impact of estrogens on human, animal and plant life: a critical review. Environ Int 99:107–119
Mompelat S, Jaffrezic A, Jarde E, LeBot B (2013) Review storage of natural water samples and preservation techniques for pharmaceutical quantification. Talanta 109:31–45
Nie M, Yan C, Dong W, Liu M, Zhou J, Yang Y (2015) Occurrence, distribution and risk assessment of estrogens in surface water, suspended particulate matter, and sediments of the Yangtze Estuary. Chemosphere 127:109–116
Nie M, Yang Y, Liu M, Yan C, Shi H, Dong W, Zhou JL (2014) Environmental estrogens in a drinking water reservoir area in Shanghai: occurrence, colloidal contribution and risk assessment. Sci Total Environ 487:785–791
Sangster JL, Oke H, Zhang Y, Bartelt-Hunt SL (2015) The effect of particle size on sorption of estrogens, androgens and progestagens in aquatic sediment. J Hazard Mater 299:112–121
Yang L, Cheng Q, Lin L, Wang X, Chen B, Luan T, Tam NFY (2016) Partitions and vertical profiles of 9 endocrine disrupting chemicals in an estuarine environment: effect of tide, particle size and salinity. Environ Pollut 211:58–66
Petrovic M (2014) Review methodological challenges of multi-residue analysis of pharmaceuticals in environmental samples. Trends Anal Chem 1:25–33
Ademollo N, Patrolecco L, Polesello S, Valsecchi S, Wollgast J, Mariani G, Hanke G (2012) The analytical problem of measuring total concentrations of organic pollutants in whole water. Trends Anal Chem 36:71–81
Neale PA, Pronk W, Schaëfer AI (2009) Influence of pH on losses of analyte estradiol in sample prefiltration. Environ Eng Sci 26:1157–1161
de Graaff MS, Vieno NM, Kujawa-Roeleveld K, Zeeman G, Temmink H, Buisman CJN (2011) Fate of hormones and pharmaceuticals during combined anaerobic treatment and nitrogen removal by partial nitritation-anammox in vacuum collected black water. Water Res 45:375–383
Erger C, Schmidt TC (2014) Disk-based solid-phase extraction analysis of organic substances in water. Trends Anal Chem 61:74–82
EN 16691 (2015) Water quality—determination of selected polycyclic aromatic hydrocarbons (PAH) in whole water samples—method using solid phase extraction (SPE) with SPE-disks combined with gas chromatography mass spectrometry (GC–MS), February 2015, AFNOR, La Plaine Saint Denis
EN 16693 (2015) Water quality—determination of organochlorine pesticides (OCP) in whole water samples—method using solid phase extraction (SPE) with SPE-disks combined with gas chromatography mass spectrometry (GC–MS) November 2015, AFNOR, La Plaine Saint Denis
EN 16694 (2015) Water quality—determination of selected polybrominated diphenly ether (PBDE) in whole water samples—method using solid phase extraction (SPE) with SPE-disks combined with gas chromatography–mass spectrometry (GC–MS) November 2015, AFNOR, La Plaine Saint Denis
Noppe H, De Wasch K, Poelmans S, Van Hoof N, Verslycke T, Janssen CR, De Brabander HF (2005) Development and validation of an analytical method for detection of estrogens in water. Anal Bioanal Chem 382:91–98
Česen M, Heath E (2017) Disk-based solid phase extraction for the determination of diclofenac and steroidal estrogens E1, E2 and EE2 listed in the WFD watch list by GC–MS. Sci Total Environ 590–591:832–837
Qi BL, Liu P, Wang QY, Cai WJ, Yuan BF, Feng YQ (2014) Derivatization for liquid chromatography–mass spectrometry. Trends Anal Chem 59:121–132
Bichon E, Béasse A, Prevost S, Christien S, Courant F, Monteau F, Le Bizec B (2012) Improvement of estradiol esters monitoring in bovine hair by dansylation and liquid chromatography/tandem mass spectrometry analysis in multiple reaction monitoring and precursor ion scan modes. Rapid Commun Mass Spectrom 26:819–827
Lien GW, Chen CY, Wang GS (2009) Comparison of electrospray ionization, atmospheric pressure chemical ionization and atmospheric pressure photoionization for determining estrogenic chemicals in water by liquid chromatography tandem mass spectrometry with chemical derivatizations. J Chrom A 1216:956–966
Zhang F, Bartels MJ, Brodeur JC, McClymont EL, Woodburn KB (2004) Quantitation of 17α-ethinylestradiol in aquatic samples using liquid–liquid phase extraction, dansyl derivatization, and liquid chromatography/positive electrospray tandem mass spectrometry. Rapid Commun Mass Spectrom 18:2739–2742
AFNOR, NF T 90-210, Water quality—protocol for the initial method performance assessment in a laboratory, La Plaine Saint-Denis, France, May 2009
ISO 11352 Water quality—estimation of measurement uncertainty based on validation and quality control data, La Plaine Saint-Denis, France, February 2012
Capdeville MJ, Budzinski H (2011) Trace-level analysis of organic contaminants in drinking waters and groundwaters. Trends Anal Chem 30:586–606
Devier MH, Le Menach K, Viglino L, Di Gioia L, Lachassagne P, Budzinski H (2013) Ultra-trace analysis of hormones, pharmaceutical substances, alkylphenols and phthalates in two French natural mineral waters. Sci Total Environ 443:621–632
Esteban S, Gorga M, Petrovic M, González-Alonso S, Barceló D, Valcárcel Y (2014) Analysis and occurrence of endocrine-disrupting compounds and estrogenic activity in the surface waters of Central Spain. Sci Total Environ 466–467:939–951
Avar P, Zrínyi Z, Maász G, Takátsy A, Lovas S, Tóth LG, Pirger Z (2016) β-Estradiol and ethinyl-estradiol contamination in the rivers of the Carpathian Basin. Environ Sci Pollut Res 23:11630–11638
Tavazzi S, Mariani G, Comero S, Ricci M, Paracchini B, Skejo H, Gawlik BM (2016) Water framework directive watch list method analytical method for the determination of compounds selected for the first surface water watch list. EUR 27813EN
Loos R, Gawlik BM, Locoro G, Rimaviciute E, Contini S, Bidoglio G (2009) EU-wide survey of polar organic persistent pollutants in European river waters. Environ Pollut 157:561–568
MUKIT. http://www.syke.fi/en-us/Services/Calibration_services_and_contract_laboratory/MUkit__Measurement_Uncertainty_Kit. Accessed 10 Apr 2018
Acknowledgements
This work was supported by the French Agency for Biodiversity (AFB) and national reference laboratory for monitoring aquatic Environments (AQUAREF). The authors acknowledge the six French water agencies for sampling.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lardy-Fontan, S., Le Diouron, V., Fallot, C. et al. Toward the determination of estrogenic compounds in the framework of EU watch list: validation and implementation of a two-step solid phase extraction–liquid phase chromatography coupled to tandem mass spectrometry method. Accred Qual Assur 23, 285–295 (2018). https://doi.org/10.1007/s00769-018-1346-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00769-018-1346-4