
Abstract
Eclipta alba and Guiera senegalensis have traditionally been used in Ayurvedic and Unani medicine to cure several ailments. Due to the high demand, the quantitative standardization of plant-based products is challenging to maintain their quality. We aimed to develop and validate a high-performance thin-layer chromatography (HPTLC) method for the simultaneous detection and quantification of gallic acid and quercetin in E. alba and G. senegalensis, respectively. Aqueous extracts of both plants were prepared. Qualitative and quantitative analyses were performed for the simultaneous separation of gallic acid and quercetin in each extract as per the International Conference on Harmonization (ICH) guidelines with respect to linearity, specificity, accuracy, precision, limit of detection (LOD), limit of quantification (LOQ) and robustness. HPTLC quantitative analyses for E. alba aqueous extracts revealed 3.83 µg/mg of gallic acid and 2.26 µg/mg quercetin, while 4.71 µg/mg of gallic acid and 1.06 µg/mg of quercetin for G. senegalensis. LOD was found to be 3.30 ng/band and 12.70 ng/band, while LOQ was found to be 10.20 and 38.60 ng/band for gallic acid and quercetin, respectively. The HPTLC method showed good linearity, recovery and robustness of the validated markers. This developed validated method is capable of quantifying and resolving gallic acid and quercetin and the quality control of E. alba and G. senegalensis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Medicinal plants possess a variety of bioactive compounds with important pharmacological properties, and their extracts and fractions have been used for treating many diseases since ancient times [1]. In the recent past and present, the use of herbal medicine has become increasingly popular and gaining importance because of the vast chemical diversity that they offer in both developed and developing countries, and a need has been felt for ensuring quality, safety, and efficacy of herbal drugs [2]. Herbal medicines play an important role in the health management and prevention of diseases. With the increasing need for safer medicines or drugs, herbal medicines have been enormously preferred to manage and cure human diseases because of the ease of accessibility and most importantly low toxicity [3].
Eclipta alba is commonly found in Asian countries and has traditionally been used in Ayurvedic and Unani medicine for the management of various diseases [4]. This plant has been experimentally proven to possess nephroprotective, hepatoprotective, anticancer, antiasthma features, mosquito larvicidal and toxicity against the hepatitis C virus, hair growth-promoting activity, immune-modulatory property, antioxidant, lipid-lowering activity and antivenom property [5, 6]. Guiera senegalensis is one of the most important Asian and West African medicinal plants, often used to treat many complications such as respiratory and gastrointestinal disorders, microbial and parasitic infections [7].
Among several groups of phytoconstituents, polyphenols are frequently found in fruits, vegetables, grains, bark, roots, stems, leaves, flowers, etc. [8]. Gallic acid and quercetin are some of the most important polyphenols present in a wide variety of medicinal plants including E. alba and G. senegalensis and possess a wide variety of pharmacological actions, being well known for their beneficial effects on health. Gallic acid and its derivatives are phenolic acids of plant metabolites widely spread throughout the plant kingdom [9]. Several beneficial effects are reported for gallic acid, including antioxidant, anti-inflammatory and antineoplastic properties. This compound has been reported to have therapeutic activities in gastrointestinal, neuropsychological, metabolic and cardiovascular disorders, which make it a significant option to be used as therapeutic agent or dietary supplement [10]. Quercetin is classified as a flavonol, which is one of the six sub-categories of flavonoid compounds and is the major polyphenolic flavonoid found in various vegetables and fruits [11]. Quercetin is one of the most important plant molecules that has shown many pharmacological activities, such as being anticancer, antiviral, and treating allergic, metabolic, and inflammatory disorders, eye and cardiovascular diseases and arthritis [12].
The qualitative and quantitative analyses of bioactive metabolites present in medicinal plants are one of the biggest challenges faced by phytochemists [13]. Therefore, to overcome these challenges, WHO (World Health Organization) emphasizes the various chromatographic and spectroscopic techniques such as high-performance liquid chromatography (HPLC), high-performance thin-layer chromatography (HPTLC), liquid chromatography‒mass spectrometry (LC‒MS), gas chromatography‒mass spectrometry (GC‒MS), nuclear magnetic resonance (NMR), etc., for documentation and assessment of the quality, safety and efficacy of plant-based medicines [14].
HPTLC is one of the sophisticated instrumental techniques based on the full capabilities of thin-layer chromatography. Its advantages are automation, scanning, full optimization, selective detection principle, minimum sample preparation, hyphenation, and it has emerged as a simple, reliable and efficient method not only for the chromatographic information of complex mixtures of pharmaceuticals, natural products, clinical samples, food stuffs but also for stability, dissolution or content uniformity of raw materials and formulated products [15].
Thus, to address these issues, we have developed a HPTLC method for the simultaneous determination of gallic acid and quercetin simultaneously in E. alba and G. senegalensis. The proposed method was qualitatively and quantitatively validated in relation to linearity, specificity, accuracy, precision, limit of detection (LOD), limit of quantification (LOQ) and robustness as per the International Conference on Harmonisation (ICH) guidelines [16].
2 Experimental
2.1 Chemicals and plant materials
Standard gallic acid (CAS Number 149-91-7; ≥ 97.5%) and quercetin (CAS Number 117-39-5; ≥ 95%) were procured from Sigma-Aldrich (St. Louis, MO, USA). The herbal drugs such as fresh leaves of E. alba and G. senegalensis were collected from a local market, Khari Bawli, New Delhi, India. The specimens were authenticated by Dr. Muhammad Arif, a taxonomist at the Department of Pharmacognosy and Phytochemistry, School of Pharmaceutical Sciences, Integral University, Lucknow, India. Further, voucher specimen (E. alba: 2019/No. 891; G. senegalensis: 2019/No. 892) was deposited in the Department of Pharmacognosy and Phytochemistry, Integral University, for future references. All other chemicals and reagents were of analytical grade.
2.2 Preparation of samples of E. alba and G. senegalensis
The leaves were shade-dried at room temperature for 8 days. Amounts of 10 g of the dried leaves of each plant (E. alba and G. senegalensis) were accurately weighed separately and transferred in a round-bottom flask containing water (100 mL). Thereafter, soxhalation of each plant was performed separately for 2 h at 60 °C. Then, the extracts were pooled, filtered (Whatman filter paper No. 1) and dried under reduced pressure using rotary vacuum evaporator (Buchi Type; Metrex Scientific Instruments Pvt. Ltd., New Delhi, India). The percentage yields for the obtained extracts were calculated and finally stored in an airtight container for further use.
2.3 Quantitative validation for the simultaneous separation of gallic acid and quercetin using HPTLC
Aliquots of 30 mg of each extract were dissolved individually in HPLC grade methanol and then filtered using a poly-tetrafluoroethylene (PTFE) membrane filter of 0.2 µm pore size (Millipore, Merck, Darmstadt, Germany). Thereafter, with the help of the sample-spotter Linomat V (CAMAG, Muttenz, Switzerland), aliquots of 6 μL of each sample and standards were applied simultaneously with 6 mm wide band length to pre-washed and activated silica gel 60 F254 pre-coated HPTLC plates (20 cm × 10 cm, 200 µm thickness, Merck, Darmstadt, Germany) with the nitrogen flow providing a delivery speed of 150 nL/s. Method development was executed in a CAMAG twin-trough glass chamber (20 cm × 10 cm) saturated with the mobile phase containing toluene‒ethyl acetate‒glacial acetic acid (6:3:1, V/V) as the solvent system. The plate was developed horizontally to a distance of 80 mm at room temperature (25 °C). After drying, the spots on the developed plates were visualized under visible light and then scanned under respective wavelengths (254 nm and 366 nm) by CAMAG TLC Scanner III operated using winCATS software (V 1.44, CAMAG).
2.4 Preparation of standards
A standard stock solution of gallic acid and quercetin was prepared by dissolving 1 mg of each marker in an Eppendorf tube containing one mL HPLC grade methanol and vortexed for 2‒3 min. A stock solution of gallic acid and quercetin was further diluted with methanol to give a standard solution of gallic acid and quercetin (200 µg/mL).
2.5 Validation of the method
The proposed analytical method was validated according to the ICH guidelines for parameters such as linearity, specificity, accuracy, precision, LOD, LOQ and robustness [16].
2.5.1 Linearity
The prepared standard stock solution was diluted to obtain linearity standard solutions containing gallic acid and quercetin in the concentration range of 200–2000 ng/spot, respectively. Every step was performed in triplicate. Every set was analyzed to obtain a calibration curve. The standard deviation (SD), coefficient of determination (r2), slope and intercept of the calibration curves were estimated to determine the method linearity.
2.5.2 Specificity
The specificity of the method was ascertained by analyzing standard gallic acid and quercetin. Sample solutions were applied to a TLC plate, and the plate was developed and scanned as described above. The peak purities of gallic acid and quercetin were assessed by comparing the spectra of marker compounds at three different levels, i.e., peak start, peak middle and peak end positions of the spot/bands.
2.5.3 Accuracy
Accuracy of the method was estimated through the % recoveries of known quantities of the combination of gallic acid and quercetin using the standard addition method. Known amounts of samples were spiked with known amounts of standard gallic acid and quercetin, and the % ratios between the recovered and expected concentrations were calculated. The values of % recovery and the average value of % recovery for both gallic acid and quercetin were calculated.
2.5.4 Precision
The present study was validated for intra-day and inter-day precision. Intra-day and inter-day precision was determined in triplicate by the same method on the same day for different concentrations of gallic acid (200‒2000 ng/spot) and quercetin (200‒2000 ng/spot). The repeatabilities of each sample application and calculation of the peak area for the analytes were articulated in terms of percent standard deviation (%RSD).
2.5.5 Limit of detection and limit of quantification
LOD is the lowest level of analytes that can be detected in a sample but not necessarily quantified, under the stated experimental conditions. LOQ was identified as the lowest amount of analytes that can be detected and quantified with acceptable accuracy, precision and variability. LOD and LOQ are calculated by the signal-to-noise method [17].
LOD = 3.3 × SD of obtained area /slope of the line.
LOQ = 10 × SD of obtained area / slop of the line.
2.5.6 Robustness
The determination was performed by selecting different parameters (mobile phase composition and volume and chamber saturation time) within certain limits (± 10%), and there has been no notable alteration found in the method performance and the results obtained. The results were indicated by the %RSD between the data at each variable condition.
3 Results and discussion
The resultant aqueous extracts were filtered evaporated to dryness, and the percentage yields were recorded as 9.26% and 7.39% for E. alba and G. senegalensis, respectively. Plants are considered as biosynthetic factory and produce a number of secondary metabolites like polysaccharides, alkaloids, steroids, glycosides, carbohydrates, bioflavonoids, being responsible for imparting therapeutic effects. Polyphenols are the present day panacea as they are established to exert protective action in oxidative stress-related pathologies, such as cardiovascular and metabolic diseases, cancer and a variety of neurodegenerative disorders [18]. Gallic acid and quercetin are some of the most important polyphenols that are present in a wide variety of medicinal plants including E. alba and G. senegalensis and possess a wide variety of pharmacological actions [19, 20].
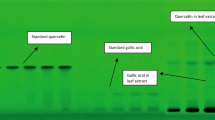
Owing to the high automatization, HPTLC technique can be used for the analysis of complex mixtures of natural products. A clear recognition by WHO and pharmacopoeias points out the importance of this technique as the method of choice for phytochemical analysis and plant identification of bioactive metabolites and impurities in herbal drugs and botanicals [21]. Therefore, the chromatographic conditions were set to obtain an analytical fingerprint containing complete information of chemical composition with a good resolution and a reasonable analysis time [22]. Based on hit and trial, different proportions of toluene‒ethyl acetate‒glacial acetic acid were tried as the mobile phase on silica gel HPTLC plates and the ratio of 6:3:1 (V/V) gave good resolution. Other chromatographic conditions, such as the sample application rate and volume, sample application positions, chamber saturation time, total run length and distance between tracks, were also optimized to give accurate, precise, and reproducible RF values, symmetrical peak shape, and better resolution for both drugs. The obtained chromatographic data revealed well-resolved and separated peaks for each metabolites at the respective wavelengths for both the samples. A typical chromatogram of E. alba and G. senegalensis at 254 nm and 366 nm, respectively, is depicted in Figs 1a, b and 2a, b. Gallic acid and quercetin appeared clearly on the HPTLC plate, scanned at 254 nm and 366 nm, respectively. Standard gallic acid (RF 0.58) and quercetin (RF 0.76) showed a single peak in HPTLC chromatogram presented in Fig. 3a, b. The calibration curve of gallic acid and quercetin was prepared by plotting concentration of gallic acid versus area of the peak and concentration of quercetin versus area of the peak, respectively, shown in Figs. 4 and 5.

HPTLC chromatogram of aqueous extract of Eclipta alba at a 254 nm and b 366 nm

HPTLC chromatogram of aqueous extract of Guiera senegalensis at a 254 nm and b 366 nm

HPTLC chromatogram of gallic acid and quercetin at a 254 nm and b 366 nm

The calibration curve of gallic acid prepared by plotting concentration of gallic acid versus peak area

The calibration curve of quercetin prepared by plotting concentration of gallic acid versus peak area
3.1 Method validation
The developed HPTLC method for the estimation of gallic acid and quercetin was validated using the following parameters such as linearity, specificity, accuracy, precision, LOD, LOQ and robustness according to ICH guidelines [16].
3.1.1 Linearity
Linearity was analyzed through the standard plot of gallic acid and quercetin. The developed method was linear in the concentration range of 200‒2000 ng with correlation coefficients of 0.9915 ± 0.011 and 0.9907 ± 0.013, respectively, when the peak area of the resolved band was plotted against concentration, thus exhibiting good linearity between concentration and peak area as shown in Table 1.
3.1.2 Specificity
The developed validated method for simultaneous separation of gallic aid and quercetin was found to be specific, as no interfering or contaminating peaks were detected. It was also evident from the peak purity data.
3.1.3 Precision
The proposed method was found to be precise as indicated by intermediate precision studies expressed as %RSD for intra-day and inter-day variations. The experimental outcome showed 0.047‒0.346 and 0.046‒0.394%RSD on inter-day precession, while 0.063‒0.185 and 0.062‒0.588%RSD on intra-day precession for gallic acid and quercetin, respectively, which suggests the validated method being precise as shown in Table 2.
3.1.4 Accuracy
The proposed method used for the quantification of marker after spiking with the standard afforded recovery of gallic acid and quercetin in the range of 98.91%–113.16% and 100.75%–113.675%, respectively, shown in Table 3.
3.1.5 Limit of detection and limit of quantification
The LOD values for gallic and quercetin acid were found to be 3.30 ng/band and 12.70 ng/band, while the LOQ values were found to be 10.20 ng/band and 38.60 ng/band, respectively, for gallic acid and quercetin, presented in Table 4. LOD and LOQ are deliberated by the method (signal to noise) according to the ICH recommendations. The method also passed the robustness test. These results indicated that the method is quite robust, accurate, precise and specific for the HPTLC analysis of gallic acid and quercetin.
3.1.6 Robustness
The parameters of the optimized methods were intentionally varied with respect to solvent system, tank saturation time, effective wavelength and observed peak area. Further, the analytical changes for both drugs were examined statistically. The summary of the robustness is shown in Table 4. The values of the %RSD of the peak areas for both gallic acid and quercetin in the analysis did not exceed the optimum range. The satisfactory low values of %RSD of peak areas and the unaffected RF values suggest the robustness of the method proposed.
3.2 Quantitative estimation of markers in extracts
In the extract of E. alba, gallic acid and quercetin were found as 3.83 μg/mg and 2.26 μg/mg, respectively, whereas in the extract of G. senegalensis, gallic acid and quercetin were found as 4.71 μg/mg and 1.06 μg/mg, respectively.
In summary, a HPTLC method was developed for the simultaneous quantification of gallic acid and quercetin, respectively, in E. alba and G. senegalensis, on the basis of their chromatographic separation and retention. In order to show its reliability, this method will be applied to different plant extracts or formulations. The comparison of the quantification of two major metabolites (gallic acid and quercetin) with HPTLC in these plants showed good correlations.
4 Conclusion
The present study illustrates a simple, accurate and precise HPTLC method for the simultaneous estimation of gallic acid and quercetin in E. alba and G. senegalensis. Notably, HPTLC fingerprints are implemented as a potential cost-effective, simple and highly selective tool that can ensure both quality and quantity of the constituents in these plants. This method may be useful for the development of quality control and marker analysis profile of these medicinal plants, which may be utilized for ensuring the quality control of plant raw materials for food and pharmaceutical preparations.
References
Alamgir ANM (2018) Biotechnology, in vitro production of natural bioactive compounds, herbal preparation, and disease management (treatment and prevention). Alamgir ANM Therapeutic Med Plants Extr 2:585–664. https://doi.org/10.1007/978-3-319-92387-1_7
Ekor M (2014) The growing use of herbal medicines: issues relating to adverse reactions and challenges in monitoring safety. Front Pharmacol 4:177. https://doi.org/10.3389/fphar.2013.00177
Yuan H, Ma Q, Ye L, Piao G (2016) The traditional medicine and modern medicine from natural products. Molecules 21:559. https://doi.org/10.3390/molecules21050559
Bhattacharjee A, Chakraborty A, Chakraborty S, Biswas P (2017) Standardization of Eclipta alba by HPTLC. HPLC AAS Pharm Anal Acta 8:4. https://doi.org/10.4172/2153-2435.1000541
Hoque R, Jiban SRM (2019) Phytochemical analysis and TLC studies of Eclipta alba and Scoparia dulcis plant extract-hot extraction. Int J Drug Dev Res 11:1. https://doi.org/10.36648/0975-9344.11.2.130
Jahan R, Al-Nahain A, Majumder S, Rahmatullah M (2014) Ethnopharmacological significance of Eclipta alba (L.) Hassk. (Asteraceae). Int Sch Res Notices 2014:385969. https://doi.org/10.1155/2014/385969
Dirar AI, Devkota HP (2020) Ethnopharmacological uses, phytochemistry and pharmacological activities of Guiera senegalensis J.F. Gmel. (Combretaceae). J Ethnopharmacol 267:113433. https://doi.org/10.1016/j.jep.2020.113433
Alam P, Parvez MK, Arbab AH, Al-Dosari MS (2017) Quantitative analysis of rutin, quercetin, naringenin, and gallic acid by validated RP- and NP-HPTLC methods for quality control of anti-HBV active extract of Guiera senegalensis. Pharm Biol 55(1):1317–1323. https://doi.org/10.1080/13880209.2017.1300175
Gao J, Hu J, Hu D, Yang X (2019) A role of gallic acid in oxidative damage diseases: a comprehensive review. Nat Prod Commun 8:1–9. https://doi.org/10.1177/1934578X19874174
Kahkeshani N, Farzaei F, Fotouhi M et al (2019) Pharmacological effects of gallic acid in health and diseases: a mechanistic review. Iran J Basic Med Sci 22(3):225–237. https://doi.org/10.22038/ijbms.2019.32806.7897
Anand DAV, Arulmoli R, Parasuraman S (2016) Overviews of biological importance of quercetin: a bioactive flavonoid. Pharmacogn Rev 10(20):84–89. https://doi.org/10.4103/0973-7847.194044
Dabeek WM, Marra MV (2019) Dietary quercetin and kaempferol: bioavailability and potential cardiovascular-related bioactivity in humans. Nutrients 11(10):2288. https://doi.org/10.3390/nu11102288
Atanasov AG, Waltenberger B, Pferschy-Wenzig E-M, et al. (2015) Discovery and resupply of pharmacologically active plant-derived natural products: a review. Biotechnol Adv 33(8):1582–1614. https://doi.org/10.1016/j.biotechadv.2015.08.001
Juszczak AM, Zovko-Končić M, Tomczyk M (2019) Recent trends in the application of chromatographic techniques in the analysis of luteolin and its derivatives. Biomolecules 9(11):731. https://doi.org/10.3390/biom9110731
Mukherjee D, Kumar NS, Khatua T, Mukherjee PK (2010) Rapid validated HPTLC method for estimation of betulinic acid in Nelumbonucifera (Nymphaeaceae) rhizome extract. Phytochem Anal 21(6):556–560. https://doi.org/10.1002/pca.1232
ICH, (2005) Validation of Analytical Procedures: Text and Methodology, Q2 (R1), International Conference on Harmonization, Geneva.
Patel NG, Patel KG, Patel KV, Gandhi TR (2015) Validated HPTLC method for quantification of luteolin and apigenin in Premna mucronata Roxb Verbenaceae. Adv Pharmacol Sci 2015:682365. https://doi.org/10.1155/2015/682365
Vauzour D, Rodriguez-Mateos A, Corona G, Oruna-Concha MJ, Spencer JP (2010) Polyphenols and human health: prevention of disease and mechanisms of action. Nutrients 2(11):1106–1131. https://doi.org/10.3390/nu2111106
Yadav NK, Arya RK, Dev K, Sharma C, Hossain Z, Meena S, Arya KR, Gayen JR, Datta D, Singh RK (2017) Alcoholic extract of Eclipta alba shows in vitro antioxidant and anticancer activity without exhibiting toxicological effects. Oxid Med Cell Longev 2017:9094641. https://doi.org/10.1155/2017/9094641
Dirar AI, Alsaadi DHM, Wada M, Mohamed MA, Watanabe T, Devkota HP (2019) Effects of extraction solvents on total phenolic and flavonoid contents and biological activities of extracts from Sudanese medicinal plants. South African J Bot 120:261–267. https://doi.org/10.1016/j.sajb.2018.07.003
Upadhye AS, Rajopadhye A, Dias A (2018) Development and validation of HPTLC fingerprints of three species of Alpinia with biomarker Galangin. BMC Complement Altern Med 18(1):16. https://doi.org/10.1186/s12906-017-2033-4
Donno D, Boggia R, Zunin P et al (2016) Phytochemical fingerprint and chemometrics for natural food preparation pattern recognition: an innovative technique in food supplement quality control. J Food Sci Technol 53:1071–1083. https://doi.org/10.1007/s13197-015-2115-6
Acknowledgements
This project was supported by a Research Grant from the Deanship of Scientific Research, Prince Sattam Bin Abdulaziz University, Alkharj, Kingdom of Saudi Arabia (Grant No. 2020/03/17251).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Khalid, M., Alqarni, M.H., Foudah, A.I. et al. A high-performance thin-layer chromatography method for the simultaneous determination of quercetin and gallic acid in Eclipta alba and Guiera senegalensis. JPC-J Planar Chromat 34, 71–77 (2021). https://doi.org/10.1007/s00764-021-00084-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00764-021-00084-x