
Abstract
We succeeded in expressing selenocysteine β-lyase (SCL) from a lactic acid bacterium, Leuconostoc mesenteroides LK-151 (Lm-SCL), in the soluble fractions of Escherichia coli Rosetta (DE3) using a novel expression vector of pET21malb constructed by ourselves that has both maltose binding protein (MBP)- and 6 × His-tag. Lm-SCL acted on l-selenocysteine, l-cysteine, and l-cysteine sulfinic acid but showed a high preference for l-selenocysteine. The kcat and kcat/Km values of Lm-SCL were determined to be 108 (min−1) and 42.0 (min−1・mM−1), respectively, and this was enough catalytic efficiency to suggest that Lm-SCL might also be involved in supplying elemental selenium from l-selenocysteine to selenoproteins like other SCLs. The optimum temperature and optimum pH of Lm-SCL were determined to be 37 °C and pH 6.5, respectively. Lm-SCL was stable at 37–45 °C and pH 6.5–7.5. Lm-SCL was completely inhibited by the addition of hydroxylamine, semicarbazide, and iodoacetic acid. The enzyme activity of Lm-SCL was decreased in the presence of various metal ions, especially Cu2+. The quaternary structure of Lm-SCL is a homodimer with a subunit molecular mass of 47.5 kDa. The similarity of the primary structure of Lm-SCL to other SCLs from Citrobacter freundii, Escherichia coli, humans, or mouse was calculated to be 47.0, 48.0, 12.5, or 24.0%, respectively. Unlike Ec-SCL, our mutational and molecular docking simulation studies revealed that C362 of Lm-SCL might also catalyze the deselenation of l-selenocysteine in addition to the desulfuration of l-cysteine.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Selenocysteine β-lyase (SCL; EC 4.4.1.16) is classified as a Fold Type I- pyridoxal 5´-phosphate (PLP) enzyme (Eliot and Kirsch 2004) and catalyzes the conversion of l-selenocysteine into l-alanine and elemental selenium in the presence of PLP as a cofactor. SCL was first discovered in pig liver in 1982 by Professor Emeritus of Kyoto University, Dr. Kenji Soda and his colleagues (Esaki et al. 1982) and subsequently in Citrobacter freundii (Chocat et al. 1985), humans (Daher and Van 1992), Escherichia coli (Mihara et al. 1999), and Mus musculus (Mihara et al. 2000a, b). Mammalian SCLs are highly specific for l-selenocysteine, while bacterial SCLs, including NIFS-like proteins (Flint 1996; Zeng et al. 1993, 1994; Mihara et al. 1999), act on l-cysteine and l-selenocysteine but show a high preference for l-selenocysteine over l-cysteine. The functions of both mammalian and bacterial SCLs are considered to be involved in supplying elemental selenium produced from l-selenocysteine to the selenoproteins and elemental sulfur produced from l-cysteine for the formation of Fe-S complexes in organisms (Lacourciere and Stadtman 1998; Kurokawa et al. 2011). A few SCLs have been crystallized, and X-ray crystallographic analysis was performed (Mihara et al. 1999; Omi et al. 2011; Collins et al. 2012; Johansson et al. 2012), but the reaction mechanism of SCL in detail is still ambiguous. SCL is a rare enzyme in nature, especially in microorganisms, and accordingly, SCL from lactic acid bacteria has not been purified and characterized thus far. During the course of our whole-genome sequence analysis of Leuconostoc mesenteroides LK-151, which was isolated from a Japanese sake cellar as a high producer of D-amino acids (Kato and Oikawa 2017), we found that the homologous gene of SCL existed interestingly in the genome of L. mesenteroides LK-151 (accession no.: AP017936).
In this manuscript, we describe the first discovery of selenocysteine β-lyase from a lactic acid bacterium, Leuconostoc mesenteroides LK-151 (Lm-SCL), and its enzymological properties and propose its catalytic mechanism based on mutational and docking simulation studies.
Materials and methods
Materials
Leuconostoc mesenteroides LK-151 isolated from Japanese sake cellar was kindly supplied by Kiku-Masamune Sake Brewing Co., Ltd., Hyogo, Japan. Escherichia coli Rosetta (DE3) and the pET21b vector were purchased from Novagen (Merck KGaA, Darmstadt, Germany). Escherichia coli NEB10β, pMAL-c5x vector, and T4 DNA ligase were purchased from New England BioLabs (Massachusetts, USA). l-Selenocystine (purity: > 97.0%) was purchased from Tokyo chemical Ind. Co., Ltd. (Tokyo, Japan). Protein assay CBB solution was purchased from Nacalai Tesque, Inc. (Tokyo, Japan). Ni–NTA agarose was purchased from QIAGEN (Hilden, Germany). l-Alanine dehydrogenase from Bacillus stearothermophilus was obtained from Nipro Corp. (Osaka, Japan). All other reagents were the best commercially available grade and purchased from Invitrogen Co. (Carlsbad, CA, USA), Kanto Chemical Co., Inc. (Tokyo, Japan), Kishida Chemical Co., Ltd. (Tokyo, Japan), Nacalai Tesque, Inc. (Kyoto, Japan), Sigma–Aldrich Co. (St. Louis, MO, USA), Tokyo chemical Ind. Co., Ltd. (Tokyo, Japan), FUJIFILM Wako Pure Chemical Corp. (Osaka, Japan), or Watanabe Chemical Ind., unless otherwise stated.
Expression of the selenocysteine β-lyase homologous gene from Leuconostoc mesenteroides LK-151
First, we constructed a new expression vector using pMAL-c5x and a pET21b vector that has both maltose binding protein (MBP)- and 6XHis-tags to express the selenocysteine β-lyase homologous gene from Leuconostoc mesenteroides LK-151 (Lm-scl) in the soluble fractions of E. coli Rosetta (DE3) cells under the control of the T7-lac promoter and purified the enzyme unless otherwise stated. From the pMAL-c5x vector, the coding region of the MBP-tag together with its ribosome binding site and factor Xa protease recognition sequence (malE cassette) was amplified by PCR using a set of primers as follows: MALcassette-xbaF, 5’-ATTCCCCTCTAGAATTTTCACGAGCAATTGACCAAC-3’; and MALcassette-ndeR, 5’-TGCTAGCCATATGTGAAATCCTTCCCTCGATC-3’. Underlined regions represent restriction sites for subsequent cloning. The resulting 1.2-kb fragment was digested with NdeI and XbaI and ligated with a similarly digested pET21b plasmid, yielding an MBP-tag fused protein expression vector, pET21malb, in which the malE cassette was placed between the T7-lac promoter and the multiple cloning site.
L. mesenteroides LK-151 was cultivated at 30 °C in MRS medium, and its genomic DNA was extracted using the boiling method (Sambrook et al. 1989). The Lm-scl gene was amplified by PCR from the genomic DNA of L. mesenteroides LK-151 using a pair of primers with NheI and HindIII restriction sites at the 5’ and 3’ termini of the gene, respectively: 5’-TTCATATGAACTATAATGCACCACA-3’ and 5’-AACTCGAGTGAAACAATTATTCGT-3’. The purified PCR product and a pET21malb vector were double digested with the restriction enzymes NheI and HindIII and ligated using a T4 DNA ligase.
The DNA sequence of the pET21malb-Lm-scl vector constructed was confirmed, and competent Escherichia coli Rosetta (DE3) cells were transformed with the vector using an electroporation Gene pulser Xcell™ (Bio–Rad Laboratories, Inc., California, USA) to form pET21malb-Lm-scl/Escherichia coli Rosetta (DE3). The transformant obtained was cultivated in LB medium (50 mL) in a Sakaguchi flask (500 mL) containing 0.4% (w/v) glucose and 100 μg/mL carbenicillin at 37 °C until the optical density at 600 nm was reached at approximately 0.6, and Lm-SCL was expressed by the addition of isopropyl β-d-1-thiogalactopyranoside (0.2 mM final concentration) and further cultivation at 18 °C for 18 h.
Purification of selenocysteine β-lyase homolog from Leuconostoc mesenteroides LK-151 expressed in Escherichia coli Rosetta (DE3)
pET21malb-Lm-scl/Escherichia coli Rosetta (DE3) cells were collected by centrifugation at 7190×g for 15 min at 4 °C using a Centrifuge 5430R (Eppendorf, Tokyo, Japan) and suspended in 50 mM sodium phosphate buffer (NaPB), pH 8.0, containing 300 mM NaCl (buffer A) plus 10 mM imidazole (lysis buffer). This cell suspension was transferred to a beaker PYREX® (10 mL) and treated with a sonicator (UD-201, Tomy Seiko, Tokyo) in ice-cooled water to disrupt the cells (output, 6; duty cycle, 50%). After ultrasonication, the suspension was centrifuged at 7,190×g for 15 min at 4 °C. The supernatant was transferred into a centrifuge tube (15 mL) (Thermo Scientific, Tokyo) to remove insoluble materials, and the supernatant was used as a cell-free extract. The cell-free extract was then applied to a column of Ni–NTA agarose (φ1.8 × 2.0 cm) (Qiagen, Hilden, Germany) that had been equilibrated with lysis buffer. The unabsorbed proteins were washed with buffer A plus 20 mM imidazole. The adsorbed protein (MBP-Lm-SCL) was eluted with 8 mL of buffer A plus 250 mM imidazole then collected and stored as a purified enzyme at − 80 °C after it was dialyzed against 50 mM potassium phosphate buffer (KPB), pH 7.0, containing 10% (v/v) glycerol. The purified MBP-Lm-SCL was treated with factor Xa to cleave the MBP-tag, if necessary. The reaction mixture (total volume: 50 μL) containing MBP-Lm-SCL (10 μg), Factor Xa (1 μg/μL, diluted with dilution/storage buffer (Novagen); 1 μL), Factor Xa cleavage/capture buffer (5 μL), and deionized water was incubated at 20 °C for 18 h.
Standard assay conditions
Selenocysteine β-lyase activity was assayed spectrophotometrically by measuring the amount of l-alanine produced in a reaction mixture with alanine dehydrogenase in the presence of NAD+ according to the modified method of the manufacturer: https://www.nipro.co.jp/business/others/enzymes/nipro_enzyme_201604.pdf. The standard reaction mixture (total volume: 0.1 mL) contained 1.25 mM l-selenocysteine, 80 mM KPB (pH 6.5), 0.2 mM pyridoxal-5’-phosphate, and enzyme solution. l-Selenocysteine in the reaction mixture was prepared by reduction of l-selenocystine with DTT as follows: 12.5 mM l-selenocystine dissolved in 0.3 mL of 10 mM Tris–HCl buffer, pH 8.0, containing 0.1 M DTT was incubated at 37 °C for 60 min. The reduction of l-selenocystine to l-selenocysteine was confirmed by thin-layer chromatography (developing solvent, n-butyl alcohol: acetic acid: H2O = 4:1:1; Rf values, l-selenocystine (0.08); l-selenocysteine (0.58)). The enzyme reaction was started by the addition of an enzyme solution containing 10 μg of purified enzyme unless otherwise stated and carried out at 37 °C for 0, 5, or 10 min. The enzyme reaction was terminated by heat treatment at 100 °C for 10 min. Then the reaction mixture (80 μL) was added to the second reaction mixture (total volume: 0.19 mL) containing 120 mM Gly-NaOH (pH 10.5) 70 μL, 10 mM NAD+ 20 μL, and 3 U/mL l-alanine dehydrogenase 20 μL. The reaction mixture was incubated at 37 °C, and after 30 min, the absorption at 340 nm was measured using spectrophotometer UV-1800 (Shimadzu Co. Ltd, Kyoto, Japan). One unit of selenocysteine β-lyase activity is defined as the amount of enzyme that produces 1 μmol of l-alanine per min. Before carrying out the enzyme assay under various conditions, we performed preliminary experiments repeatedly to establish the optimum conditions for each enzyme assay. All calibration curves of the concentration of l-alanine (0.0–1.0 mM) versus the absorption at 340 nm used were linear with a correlation coefficient of between 0.95 and 0.99. Protein concentration was measured by the Bradford method using Protein Assay CBB solution with bovine serum albumin as a standard. All calibration curves of the concentration of protein (0–0.5 mg/mL) versus the absorption at 595 nm used were linear with a correlation coefficient of 0.99 or 1.00. The enzyme activity measured under optimal conditions and the protein concentrations were determined in two separate experiments for any given time point (this is indicated as n = 2) for every new batch of freshly purified MBP-Lm-SCL. The average value and the standard error were calculated and are shown with the equation of calibration curve and its correlation coefficient.
Basic enzymological properties
The effects of temperature and pH on MBP-Lm-SCL activity were examined by measuring the enzyme activity under various temperature and pH conditions. The reaction temperatures were 20, 30, 37, 40, 50, or 60 °C. The buffers used were as follows: MES (pH 5.5, 6.0, and 6.5); HEPES (pH 6.5, 7.0, 7.5, and 8.0); and TAPS (pH 8.0, 8.5, and 9.0).
The thermal and pH stabilities of MBP-Lm-SCL were examined by measuring the residual activity under standard assay conditions after heat or pH treatment. Heat treatment was carried out by incubating 200 μL of purified Lm-SCL (0.46 U/mg: mean of 0.47 and 0.45 U/mg) dissolved in 50 mM KPB (pH 7.0) at 37, 45, 50, 55, or 60 °C for 60 min. The pH treatment was carried out by mixing 100 μL of purified Lm-SCL (0.49 U/mg: mean of 0.48 and 0.49 U/mg) dissolved in 50 mM KPB (pH 7.0) with an equal amount of 100 mM buffer, and the mixture was incubated at 4 °C for 24 h. The buffers used were as follows: acetate (pH 5.0 and 5.5); MES (pH 5.5, 6.0 and 6.5); HEPES (pH 6.5, 7.0, 7.5 and 8.0); and TAPS (pH 8.0, 8.5 and 9.0).
The substrate specificity of MBP-Lm-SCL for various l-selenocysteine analogs was examined under standard assay conditions. The substrates used were l-selenocysteine, dl-selenocysteine, l-cysteine, and l-cysteine sulfinic acid.
Molecular mass and quaternary structure
The subunit molecular mass of MBP-Lm-SCL was determined using SDS–PAGE with a 10% T polyacrylamide gel. The purified MBP-Lm-SCL (20 μL) was mixed with 2 × sample buffer (20 μL) containing 125 mM Tris–HCl (pH 6.8), 4% (w/v) SDS, 10% (w/v) sucrose, 0.01% (w/v) bromophenol blue, and 5% (w/v) ( ±)-dithiothreitol, and the mixture was then incubated at 95 °C for 3 min. The molecular mass of MBP-Lm-SCL was estimated by gel filtration. The standard proteins used were blue dextran (2000 kDa), thyroglobulin (669 kDa), ferritin (440 kDa), aldolase (158 kDa), conalbumin (75 kDa), and ovalbumin (44 kDa). Each standard protein (5.0 mg) was dissolved in 0.1 mL of Milli-Q, and the mixture was applied to a Superdex 200 column (φ10 × 300 mm, GE Health care, Tokyo, Japan) equilibrated with 50 mM KPB (pH 7.0). The column was eluted with the same buffer at a flow rate of 0.5 mL/min. Purified MBP-Lm-SCL (1.5 mg/ml, 0.1 mL) was applied to the column under the same conditions.
Kinetic analysis
Lm-SCL activity was measured with various concentrations of l-selenocysteine (0.50, 0.75, 1.00, and 1.25 mM) under standard assay conditions. The kinetic parameters (Km and kcat) of the reaction of MBP- Lm-SCL with l-selenocysteine were determined by analyzing the enzyme activities with Lineweaver–Burk plots (Lineweaver and Burk 1934).
Effect of inhibitors
The effects of various inhibitors on MBP-Lm-SCL were determined as follows: the enzyme solution (1.0 mg/mL) dissolved in 50 mM KPB (pH 7.0) + 5 mM DTT was incubated at 37 °C for 30 min with 1.0 or 10 mM of various reagents, such as l-, d-, dl-penicillamine, hydroxylamine, semicarbazide, and iodoacetic acid, and the residual activity of MBP-Lm-SCL was measured under standard assay conditions.
Effects of metal ions and reagents on enzyme activity
To determine the effect of metal ions and reagents on the activity of MBP-Lm-SCL, we measured the enzyme activity in the presence of various metal ions and reagents (final concentration: 1 mM), such as PbCl2, ZnCl2, NiCl2, FeCl2, CuCl2, MnCl2, CoCl2, HgCl2, p-chloromercuribenzoic acid, and ethylenediaminetetraacetic acid. MBP-Lm-SCL (1.12 U/mg, 180 μL) dissolved in 50 mM KPB (pH 7.0) was mixed with 20 μL of 10 mM metal ion or SH-reagent solution, and then the mixture was incubated at 4 °C for 60 min. After dialysis against 50 mM KPB (pH 7.0), the residual activity was measured under standard assay conditions.
Spectroscopic analysis
The absorption spectra of MBP-Lm-SCL were measured using a Hitachi U-3900H spectrophotometer at room temperature (approximately 25 °C). The holo form of MBP-Lm-SCL was prepared after dialysis of the purified MBP-Lm-SCL against 50 mM KPB, pH 7.0 (buffer B), for 16 h at 4 °C. The apo form of the enzyme was prepared after dialysis of the purified MBP-Lm-SCL against buffer B containing 10 mM hydroxylamine at 4 °C for 16 h. The reduced form of the enzyme was prepared after dialysis of the purified MBP-Lm-SCL against buffer B containing 8 mM sodium borohydride at 4 °C for 16 h.
Molecular docking of L-selenocysteine or L-cysteine to Lm-SCL
The Modeller program (ver. 9.23; https://salilab.org/modeller/9.23/release.html) was used for homology modeling of Lm-SCL with a crystal structure of E. coli SufS (PDB ID: 5DB5) as a template. Molecular docking of l-selenocysteine or l-cysteine to the homology model of Lm-SCL constructed was performed using the AutoDock Vina program (https://vina.scripps.edu). The grid box around the Schiff base of chain A was set to 24 × 50 × 24 Å. The ligand molecules and side chains of H119, H120, N171, D196, Q199, S219, R357, H360, H361, C362, and R377 of chain A and H51, E250, and T273 of chain B were allowed to be flexible during the simulation.
Analysis of protein sequence alignment
Analysis of protein sequence alignment was carried out using Jalview (https://www.jalview.org) (Waterhouse et al. 2009). The secondary structure was predicted via a jury network (Cuff and Barton 1999; Cole et al. 2008).
Site-directed mutagenesis
The amino acid residues H119, E250, C362, and R377, which are candidate catalytic residues of Lm-SCL predicted from the protein sequence alignment and the molecular docking studies, were replaced singly or doubly with an alanine residue to produce H119A, C362A, R377A, H119A/C362A, and E250A/C362A-Lm-SCL by site-directed mutagenesis according to the method of Takara Bio Inc. (Kyoto, Japan). The primers used are summarized in Table 1. PCR was carried out in a reaction mixture (total volume: 20 μL) containing Prime STAR MAX Premix (10 μL), 10 μM forward primer (0.4 μL), 10 μM reverse primer (0.4 μL), pET21malb-lm-scl (< 0.1 μL), and Milli-Q (9.2 μL). The PCR mixture was transformed into Escherichia coli NEB10β, and each transformant harboring the desired mutated plasmid was cultivated and purified according to the method mentioned above. The activities of each mutated enzyme were measured under standard assay conditions.
Phylogenetic tree analysis
Phylogenetic analysis was carried out using the primary structures of Lm-SCL and 23 representative proteins homologous to Lm-SCL. The phylogenetic tree was made by the neighbor-joining method (Saitou and Nei 1987) using Protein BLAST Tree View based on the results of BLAST pairwise alignments. The analytical conditions used were as follows: max sequence difference, 0.85; and distance, Grishin (protein).
Accession number
The DNA sequence of the gene encoding the selenocysteine β-lyase and the genome from Leuconostoc mesenteroides LK-151 are available at GenBank under accession no. LC652838 and AP017396, respectively.
Results
Expression and purification of MBP-Lm-SCL
First, we tried to express Lm-scl using pET21b, but almost all of its gene product, Lm-SCL, is expressed in the insoluble fraction of E. coli BL21 (DE3) cells. Therefore, we constructed the new MBP-tag fused protein expression vector pET21malb, and using pET21malb, we succeeded in expressing MPB-Lm-SCL in a soluble fraction of E. coli BL21 (DE3) cells and purified it homogeneously (Supplemental Fig. 1). The results of purification of MBP-Lm-SCL are summarized in Table 2.
Effect of MBP-tag on the enzyme activity of Lm-SCL
We measured the enzyme activities with MPB-Lm-SCL or Factor Xa-treated MPB-Lm-SCL, that is, Lm-SCL, to confirm the effect of the MBP tag on the enzyme activity of Lm-SCL. The selenocysteine β-lyase activity of MPB-Lm-SCL (0.47 U/mg: mean of 0.45 and 0.48 U/mg) is almost same as that of Lm-SCL (0.49 U/mg: mean of 0.48 and 0.49 U/mg), and accordingly, we decided to use MPB-Lm-SCL to measure various enzymological properties of Lm-SCL except its quaternary structure. Hereafter we use the word Lm-SCL instead of MPB-Lm-SCL to describe the experiment with MPB-Lm-SCL in this manuscript.
Substrate specificity of Lm-SCL
Table 3 shows the substrate specificity of Lm-SCL. Lm-SCL acts on l-selenocysteine, l-cysteine, and l-cysteine sulfinic acid but shows a high preference for l-selenocysteine.
Basic enzymatic properties
The optimum temperature and pH of Lm-SCL were determined to be 37 °C and pH 6.5, respectively (Supplemental Fig. 2). Lm-SCL is stable between 37 and 45 °C and pH 6.5–7.5 and shows more than 60% and 50% activity, respectively, compared with the enzyme without heat or pH treatment (Supplemental Fig. 3). The treatment with hydroxylamine, semicarbazide, and iodoacetic acid completely inhibit Lm-SCL. Lm-SCL is strongly inhibited in the presence of l-, d-, and dl-penicillamine (Supplemental Table 1). The enzyme activity of Lm-SCL is decreased in the presence of various metal ions, especially Cu2+, Ca2+, Al3+, Mn2+, and Hg2+ (Supplemental Table 2). The kinetic parameters of Lm-SCL were determined and are summarized with those of other SCLs in Table 4. The quaternary structure of Lm-SCL is a homodimer with a subunit molecular mass of 47.5 kDa.
Primary structure of Lm-SCL
The primary structure of Lm-SCL was compared with that of other mammalian and bacterial SCLs previously reported (Fig. 1). The similarities between Lm-SCL and SCL from Citrobacter freundii (accession number: WP_038640267, Cf-SCL), Escherichia coli (accession number: 1JF9_A, Ec-SCL), humans (accession number: NP_057594, Human-SCL), or mouse (accession number: NP_057926, Mouse-SCL) were calculated to be 47.0, 48.0, 12.5, or 24.0%, respectively. The PLP-binding lysine residue of Lm-SCL is considered to be K222, and it is conserved between mammalian and bacterial SCLs (Fig. 1). The catalytic residues of Ec-SCL, i.e., H123, C364, and R379 are well conserved in the primary structure of Lm-SCL as H119, C362, and R377, respectively.

Alignment of the primary structure of Lm-SCL. The colors in the primary structures indicate the character of sidechain of amino acid residues and specific amino acids as follows:  : hydrophobic,
: hydrophobic,  : cationic,
: cationic,  : anionic,
: anionic,  : polar,
: polar,  : G,
: G,  : P,
: P,  : H and Y. Jnetpred: the consensus prediction. Helices are marked as red tubes, and sheets are marked as light green arrows. JNETCONF: the confidence estimates for the prediction. High values mean high confidence. JNETHMM: the HMM profile-based prediction. Helices are marked as red tubes, and sheets are marked as light green arrows. JNETPSSM: the PSSM profile-based prediction. Helices are marked as red tubes, and sheets are marked as light green arrows. JNETJURY: An asterisk in this annotation indicates that JNETJURY was invoked to rationalize significantly different primary predictions. Jnet Burial: prediction of solvent accessibility
: H and Y. Jnetpred: the consensus prediction. Helices are marked as red tubes, and sheets are marked as light green arrows. JNETCONF: the confidence estimates for the prediction. High values mean high confidence. JNETHMM: the HMM profile-based prediction. Helices are marked as red tubes, and sheets are marked as light green arrows. JNETPSSM: the PSSM profile-based prediction. Helices are marked as red tubes, and sheets are marked as light green arrows. JNETJURY: An asterisk in this annotation indicates that JNETJURY was invoked to rationalize significantly different primary predictions. Jnet Burial: prediction of solvent accessibility
Molecular docking of substrates to Lm-SCL
We performed molecular docking simulations to estimate the substrate binding of l-selenocysteine and l-cysteine molecules to the active center of Lm-SCL using the program AutoDock Vina (Fig. 2). All amino acid residues conserved in the primary structure of Lm-SCL, i.e., H119, K222, C362, and R377 exist in the active center of Lm-SCL when either l-selenocysteine or l-cysteine docked as a substrate. Accordingly, K222 is predicted strongly as the PLP-binding lysine residue of Lm-SCL by the molecular docking simulations. Interestingly, we found that E250 of Lm-SCL exists near a substrate molecule that is conserved with mammalian SCLs (human and mouse SCLs) but is replaced with a serine residue in the primary structure of bacterial SCLs (Cf- and Ec-SCLs) (Fig. 1). The side chain of E250 of Lm-SCL moves only when an l-cysteine molecule docks as a substrate (Fig. 2).

Molecular models of the substrate in the active center of Lm-SCL. The binding modes for Lm-SCL and l-selenocysteine (A) and l-cysteine (B) calculated by AutoDock Vina were visualized using PyMOL software. Carbon atoms of chains A and B of Lm-SCL, PLP molecules bound to K222 of chain A, and ligand molecules are shown in blue, green, yellow, and orange, respectively. Flexible residues during the simulation are shown in magenta. The binding affinities calculated from this simulation are noted below each image
Comparison of enzymatic activities and spectral features between Lm-SCL and various mutated Lm-SCLs
The SCL activities were compared using Lm-SCL and its mutated enzymes (Table 5). The SCL activity of Lm-SCL-H119A decreases dramatically (specific activity 0.07 U/mg), and this observation agrees well with Ec-SCL-H123A. The SCL activity of Lm-SCL-C362A decreased slightly (specific activity 0.76 U/mg), while Ec-SCL-C364A shows approximately 1.5 times higher SCL activity than Ec-SCL. We found that Lm-H119A/C362A shows no enzyme activity toward l-selenocysteine, but Lm-SCL-E250A/C362A shows almost the same activity as Lm-SCL-C362A. Lm-SCL-R377A also have no SCL activity similar to Ec-SCL-R379A.
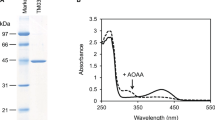
The absorption spectra of the holo-, apo-, and reduced forms of Lm-SCL were measured (Fig. 3A). The absorption maximum at 420 nm, which is derived from an internal aldimine linkage probably between K222 and PLP, is observed for Lm-SCL, and the ratio of A280 to A420 is approximately 12.8. This observation suggests that Lm-SCL binds PLP as a coenzyme. Compared with the absorption spectrum of the holo-form of Lm-SCL with that of Lm-SCL-H119A, C362A, and R377A, the absorption maximum at 420 nm decreases significantly in Lm-SCL-H119A and slightly in Lm-SCL-C362A. However, unexpectedly, in the case of Lm-SCL-R377A, the absorption maximum at 420 nm is similar to that of Lm-SCL (Fig. 3B).

Spectroscopic analysis of Lm-SCL and its mutated enzymes. A Absorption spectra of holo, apo, and reduced forms of Lm-SCL. Blue line, holo form; Green line, apo form; Red line, reduced form. The protein concentration was as follows: holo form, 0.44 mg/mL; apo form, 0.36 mg/mL; reduced form 0.37 mg/mL. The buffer used was 50 mM KPB (pH 7.0). B Comparison of the absorption spectrum of the holo-form of Lm-SCLwith that of Lm-SCL-H119A, C362A, R377A, and H119A/C362A. Blue line, Lm-SCL; Green line, Lm-SCL-C362A; Red line, Lm-SCL-H119A; Pale blue, Lm-SCL-R377A, Yellow, Lm-SCL-H119A/C362A. The protein concentration was as follows: Lm-SCL, 0.59 mg/mL; Lm-SCL-C362A, 0.54 mg/mL; Lm-SCL-H119A, 0.49 mg/mL; Lm-SCL-R377A, 0.51 mg/mL; Lm-SCL-H119A/C362A, 0.51 mg/mL. The buffer used was 50 mM KPB (pH 7.0)
Phylogenetic analysis of Lm-SCL
The Protein BLAST search shows that the homologous protein whose identity is over 62% similar to the primary structure of Lm-SCL exists in very restricted lactic acid bacteria. The phylogenetic tree of Lm-SCL based on the results of the Protein BLAST shows that the homologs of Lm-SCL form only the cluster with the proteins derived from various species of Leuconostoc, such as L. palmae, L. holzapfelii, L. carnosum, L. gelidum, L. inhae, L. miyukkimuchii, L. suionicum, L. litchi, L. pseudomesenteroides, L. falkenbergense, L. rapi, and L. fallax (Fig. 4).

Phylogenetic analysis of Lm-SCL. The phylogenetic analysis was carried out using the following sequences (accession no; identity for Lm-SCL): Leuconostoc miyukkimuchii (WP_220740652.1; 91.7%), Leuconostoc suionicum (WP_211636978.1; 82.8%), Leuconostoc litchii (WP_148606558.1; 82.4%), Leuconostoc pseudomesenteroides (TOZ08121.1; 80.6%), Leuconostoc falkenbergense (WP_188356479.1; 80.6%), Leuconostoc gelidum (WP_060391196.1; 77.5%), Leuconostoc inhae. (WP_220734553.1; 77.6%), Leuconostoc rapi (WP_204769388.1; 72.5%), Leuconostoc carnosum (WP_150280309.1; 74.0%), Leuconostoc holzapfelii (WP_168676043.1; 70.2%), Leuconostoc palmae (WP_220741874.1; 65.9%), Agrilactobacillus composti (WP_057002295.1; 63.8%), Companilactobacillus halodurans (WP_153522285.1; 64.3%), Enterococcus hulanensis (WP_206916504.1; 63.8%), Enterococcus faecalis (EGO6530340.1; 63.3%), Enterococcus gilvus (WP_221676086.1; 63.8%), Companilactobacillus heilongjiangensis (WP_041499075.1; 65.0%), Enterococcus phoeniculicola ATCC BAA-412 (EOL41599.1; 62.8%), Agrilactobacillus yilanensis (WP_125713532.1; 62.5%), Companilactobacillus huachuanensis (WP_137611559.1; 63.5%), Lentilactobacillus hilgardii (WP_003557453.1; 63.4%), and Enterococcus pseudoavium (WP_115872699.1; 64.8%)
Discussion
l-Selenocysteine β-lyase is a rare enzyme in nature, especially in bacteria. Two bacterial SCLs, namely those from Citrobacter freundii and Escherichia coli have been purified and characterized enzymologically (Chocat et al. 1985; Mihara et al. 1999). We succeeded in expressing selenocysteine β-lyase (SCL) from a lactic acid bacterium, Leuconostoc mesenteroides LK-151 (Lm-SCL), in the soluble fractions of Escherichia coli Rosetta (DE3) using a novel expression vector of pET21malb constructed by ourselves that has both maltose binding protein (MBP)- and 6 × His-tag. To our knowledge, this is the first example of SCL from a lactic acid bacterium and is also a rare example in entire organisms.
Lm-SCL acts on l-selenocysteine, l-cysteine, and l-cysteine sulfinic acid but showed a high preference for l-selenocysteine, similar to other bacterial SCLs (Table 3), and the value of the Vmax of Lm-SCL (2.58 U/mg) is similar to other bacterial SCLs, such as Ec-SCL (6.8 U/mg) and Cf-SCL (9.49 U/mg) (Table 4). The kcat/Km of Lm-SCL (42.8 min−1・mM−1) is comparable that of mouse-SCL (278.8 min−1・mM−1) (Table 4). This was enough activity to suggest that Lm-SCL might also be involved in supplying elemental selenium from l-selenocysteine to selenoproteins and elemental sulfur produced from l-cysteine for the formation of Fe-S complexes in organisms such as other SCLs (Lacourciere and Stadtman 1998; Kurokawa et al. 2011).
In the genome of L. mesenteroides LK-151 near Lm-scl (locus_tag: LEMES_00197), the genes encoding putative Fe-S cluster formation protein, NifU-like (LEMES_00196) and putative metal-sulfur cluster biosynthetic protein (LEMES_00194) existed, and whose gene products are expected to be involved in a selenium and sulfur metabolism of L. mesenteroides LK-151 and supports this hypothesis (Pilon-Smits et al. 2002).
Mihara et al. reported that C364 of Ec-SCL was not essential for the catalytic activity toward l-selenocysteine but is for l-cysteine (Mihara et al. 2002). They proposed that the reaction mechanism of Ec-SCL with l-selenocysteine is different from that with l-cysteine, and the difference is attributed to the difference in pK values between the selenohydryl group of l-selenocysteine and the thiol group of l-cysteine. The deselenation of l-selenocysteine occurs spontaneously from the ketimine intermediate of PLP and l-selenocysteine because the pK value of the selenohydryl group of l-selenocysteine (pK = 5.2) is much lower than the pH of the reaction mixture they used (pH = 7.4). However, the pK value of the thiol group of l-cysteine (pK = 8.3) is higher than pH 7.4, and the desulfuration of l-cysteine requires nucleophilic attack by C364 of Ec-SCL. However, our mutational study revealed that C362 of Lm-SCL corresponding to C364 of Ec-SCL may also catalyze the deselenation of l-selenocysteine in addition to the desulfuration of l-cysteine. The observed difference is probably attributed to the difference in pH of the assay mixture: pH 7.4 for Ec-SCL and pH 6.5 for Lm-SCL. The optimum pH of Lm-SCL (pH = 6.5) is lower than that of Ec-SCL (pH = 7.5) and is near the pK value of the selenohydryl group of l-selenocysteine. This characteristic pH profile of Lm-SCL may reveal a new aspect of the function of C362 in a bacterial SCL. Accordingly, we propose that the deselenation of l-selenocysteine catalyzed by Lm-SCL proceeds through two reaction mechanisms both with (Fig. 5A) and without (Fig. 5B) nucleophilic attack by C362 of Lm-SCL.

Proposed reaction mechanism of Lm-SCL. A Without nucleophilic attack by Cys362. B With nucleophilic attack by Cys362
Our docking simulation study showed that the positions of amino acid residues surrounding l-selenocysteine and l-cysteine molecules in the active center of Lm-SCL are not very different from each other (Fig. 2). Only the side chain position of E250 is estimated to be different, but our mutational study of E250 of Lm-SCL showed that E250 is not involved in the SCL activity of Lm-SCL. Recently, H55 in the active center of Ec-SCL studied as SufS of Escherichia coli has been confirmed to be dispensable for the catalytic activity of the desulfuration of l-cysteine by preparation of H55A mutated enzyme (Dunkle et al. 2019). H55 of Ec-SCL is also shown to be not essential for the catalytic activity of the deselenation of l-selenocysteine previously (Mihara et al. 2002). H55 of Ec-SCL forms a hydrogen bond to S254 in the active center, but the disruption of this hydrogen bond does not cause a broader structural change. Interestingly, H55 and S254 of Ec-SCL are conserved as H51 and E250 of Lm-SCL, respectively. Considering the structural similarity in the active center between Lm-SCL and Ec-SCL, the hydrogen bond is probably made between H51 and E250 of Lm-SCL, and in addition to E250 based on our mutational study, H51 is considered to be uninvolved in the SCL activity of Lm-SCL.
Several lactic acid bacteria have been reported to take up the inorganic selenium of selenous acid and convert it to elemental selenium or organic selenium (l-selenocysteine or l-selenomethionine) to accumulate Se in their cells (Kurek et al. 2016; Kousha et al. 2017; Etgeton et al. 2018). However, the selenium metabolism of lactic acid bacteria has not been clarified at all at the enzyme molecular level. Our phylogenetic analysis showed that the homologous proteins of Lm-SCL distribute in very restricted lactic acid bacteria, especially various species of Leuconostoc, and these lactic acid bacteria may metabolize inorganic and/or organic selenium. Our new finding of SCL in lactic acid bacteria aids in the elucidation of future unknown selenium metabolism of lactic acid bacteria.
Abbreviations
- DTT:
-
Dithiothreitol
- EDTA:
-
Ethylenediamine tetraacetic acid
- LB:
-
Luria–Bertani
- MRS:
-
De Man-Rogosa-Sharpe
- PCMB:
-
p-Chloromercuribenzoic acid
- PLP:
-
Pyridoxal 5´-phosphate
References
Andrew EC, Jack KF (2004) Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu Rev Biochem 73:383–415
Chocat P, Esaki N, Tanizawa K, Nakamura K, Tanaka H, Soda K (1985) Purification and characterization of selenocysteine β-lyase from Citrobacter freundii. J Bacteriol 163:669–676
Cole C, Barber JD, Barton GJ (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res 36:W197–W201
Collins R, Johansson A-L, Karlberg T, Markova N, van den Berg S, Olesen K, Hammarström M, Flores A, Schüler H, Schiavone HL, Brzezinski P, Arnér ESJ, Högbom M (2012) Biochemical discrimination between selenium and sulfur 1: a single residue provides selenium specificity to human selenocysteine lyase. ProS One 7:e30581
Cuff JA, Barton GJ (1999) Application of enhanced multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins 40:502–511
Daher R, Lente FV (1992) Characterization of selenocysteine lyase in human tissues and its relationship to tissue selenium concentrations. J Trace Elem Electrolytes Health Dis 6:1890194
Daher R, Van LF (1992) Characterization of selenocysteine lyase in human tissues and its relationship to tissue selenium concentrations. J Trace Elem Electrolytes Health Dis 6:189–194
Dunkle JK, Bruno M, Outten W, Frantom PA (2019) Structural evidence for dimer-interface driven regulation of the type II cysteine desulfurase, SufS. Biochemistry 58:687–696
Esaki N, Nakamura T, Tanaka H, Soda K (1982) Selenocysteine lyase, a novel enzyme that specifically acts on selenocysteine. mammalian distribution and purification and properties of pig liver enzyme. J Biol Chem 257:4386–4391
Etgeton HP, Altmayer T, Gonçalves TE, Schweizer YA, Oreste EQ, Ribeiro AS, Lehn DN, Volken de Souza CF, Hoehne L (2018) Assessment of selenium bioaccumulation in lactic acid bacteria. J Dairy Sci 101:10626–10635
Flint DH (1996) Escherichia coli contains a protein that is homologous in function and N-terminal sequence to the protein encoded by the NIFS gene of Azotobacter vinelandii and that can participate in the synthesis of the FeS cluster of dihydroxy acid dehydratase. J Biol Chem 271:16068–16074
Johansson A-L, Karlberg T, Collins R, Arnér ESJ, Brzezinski P, Högbom M (2012) Biochemical discrimination between selenium and sulfur 2: mechanistic investigation of the selenium specificity of human selenocysteine lyase. ProS One 7:e30528
Kato S, Oikawa T (2017) Genome sequence of Leuconostoc mesenteroides LK-151 isolated from a Japanese sake cellar as a high producer of D-amino acids. Genome Announc 30:e00661 (e717)
Kousha M, Yeganeh S, Keramat Amirkolaie A (2017) Effect of sodium selenite on the bacteria growth, selenium accumulation, and selenium biotransformation in Pediococcus acidilactici. Food Sci Biotechnol 26:1013–1018
Kurek E, Ruszczyńska A, Wojciechowski M, Łuciuk A, Michalska-Kacymirow M, Motyl I, Bulska E (2016) Bio-transformation of seleniuim in Se-enriched bacterial strains of Lactobacillus casei. Rocz Panstw Zakl Hig 67:253–262
Kurokawa S, Takehashi M, Tanaka H, Mihara H, Kurihara T, Tanaka S, Hill K, Burk R, Esaki N (2011) Mammalian selenocysteine lyase is involved in selenoprotein biosynthesis. J Nutr Sci Vitaminol 57:298–305
Lacourciere Gerard M, Stadtman Thressa C (1998) The NIFS protein can function as a selenide delivery protein in the biosynthesis of selenophosphate. J Biol Chem 273:30921–30926
Lineweaver H, Burk D (1934) The determination of enzyme dissociation constants. J Am Chem Soc 56:658–666
Mihara H, Maeda M, Fujii T, Kurihara T, Hata Y, Esaki N (1999) A nifS-like gene, csdB, encodes an Escherichia coli counterpart of mammalian selenocysteine lyase. J Biol Chem 274:14768–14772
Mihara H, Kurihara T, Watanabe T, Yoshimura T, Esaki N (2000a) cDNA cloning, purification, and characterization of mouse liver selenocysteine lyase. candidate for selenium delivery protein in selenoprotein symthesis. J Biol Chem 275:6195–6200
Mihara H, Kurihara T, Yoshimura T, Esaki N (2000b) Kinetic and mutational studies of three NifS homologs from Escherichia coli: mechanistic difference between l-cysteine desulfurase and l-selenocysteine lyase reactions. J Biochem 127:559–567
Mihara H, Fujii T, Kato S, Kurihara T, Hata Y, Esaki N (2002) Structure of externl aldimine of Escherichia coli CsdB, an IscS/NifS homolog: implications for its specificity toward selenocysteine. J BioChem 131:679–685
Omi S, Kurokawa S, Mihara H, Hayashi H, Goto M, Miyahara I, Kurihara T, Hirotsu K, Esaki N (2011) Reaction mechanism and molecular basis for selenium/sulfur discrimination of selenocysteine lyase. J Biol Chem 285:12133–12139
Pilon-Smits EAH, Garifullina GF, Abdel-Ghany S, Kato S, Mihara H, Hale KL, Burkhead JL, Esaki N, Kurihara T, Pilon M (2011) Characterization of a NifS-like chloroplast protein from Arabidopsis. Implications for its role in sulfur and selenium metabolism. Plant Physiol 130:1309–1318
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning. Cold spring harbor laboratory press, New York
Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ (2009) Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191
Zeng L, White RH, Cash VL, Jack RF, Dean DR (1993) Cysteine desulfurase activity indicates a role for NIFS in metallocluster biosynthesis. Proc Natl Acad Sci USA 90:2754–2758
Zeng L, White RH, Cash VL, Dean DR (1994) Mechanism for the desulfurization of l-cysteine catalyzed by the NIFS gene product. Biochemistry 33:4714–4720
Acknowledgements
We thank Mr. Takumi Matsumura for the construction of pET21malb-Lm-scl and detection of the SCL activity of Lm-SCL. We appreciate the contribution of Ms Chihiro Kasahara for the preparation of E250A/C362A-Lm-SCL and evaluation of its SCL activity. We thank Dr. Takashi Kimura (Nipro Corp., Osaka, Japan) for his kind gift of L-alanine dehydrogenase of Bacillus stearothermophilus.
Author information
Authors and Affiliations
Contributions
TO designed and supervised the studies. TO, KO, KY, and SK wrote first manuscript. KO and KY carried out the experiments. TO and SK performed the primary structure and phylogenetic analyses. SK carried out the molecular docking simulation. TO and SK deduced the reaction mechanism. TO wrote the revised manuscripts.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This manuscript does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling editor: S. Stuchlík.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplemental Fig. 1
SDS-polyacrylamide gel electrophoresis of Lm-SCL. Approximately 2 μg of each purified protein was analyzed by SDS-PAGE and stained with coomassie brilliant blue R-250. A 1: Lm-SCL, 2: Lm-SCL after treatment with factor Xa to cleave MBP-tag. B 1: Lm-SCL-H119A, 2: Lm-SCL-C362A, 3: Lm-SCL-R377A, 4: Lm-SCL-H119A/C362A, 5: Lm-SCL-H119A/E250A (PPTX 1240 KB)
Supplemental Fig. 2
Optimum temperature and optimum pH of Lm-SCL. A Optimum temperature. The symbols used were as follows: ●, 1st experiment; ○, 2nd experiment. B Optimum pH. The symbols (black for 1st experiment; white for 2nd experiment) used were as follows: ■, □ MES buffer; ◆, ◇ HEPES buffer; and ▲, △TAPS buffer. The calibration curves of the concentration of l-alanine (0.0–1.0 mM) versus the absorption at 340 nm used for optimum temperature and optimum pH are y = 1.15x (R2 = 0.97) and y = 0.78x (R2 = 0.95), respectively. The calibration curve of the concentration of protein (0–0.5 mg/mL) versus the absorption at 595 nm used was y = 0.95x (R2 = 1.00). Data obtained from two separate experiments under optimum conditions established based on preliminary experiments and its mean are shown. (PPTX 52 KB)
Supplemental Fig. 3
Thermal and pH stability of Lm-SCL. A Thermal stability. The symbols used were as follows: ●, 1st experiment; ○, 2nd experiment. The calibration curve of the concentration of l-alanine (0.0–1.0 mM) versus the absorption at 340 nm used is y = 0.78x (R2 = 0.97). The calibration curve of the concentration of protein (0–0.5 mg/mL) versus the absorption at 595 nm used was y = 0.99x (R2 = 0.99). Data obtained from two separate experiments under optimum conditions established based on preliminary experiments and its mean are shown. B pH stability. The symbols (black for 1st experiment; white for 2nd experiment) used were as follows: ●,○ acetate buffer; ■, □ MES buffer; ◆, ◇ HEPES buffer; and ▲, △TAPS buffer. The calibration curve of the concentration of l-alanine (0.0–1.0 mM) versus the absorption at 340 nm used is y = 1.41x (R2 = 0.99). The calibration curve of the concentration of protein (0–0.5 mg/mL) versus the absorption at 595 nm used was y = 1.00x (R2 = 1.00). Data obtained from two separate experiments under optimum conditions established based on preliminary experiments and its mean are shown (PPTX 46 KB)
Rights and permissions
About this article

Cite this article
Oikawa, T., Okajima, K., Yamanaka, K. et al. First enzymological characterization of selenocysteine β-lyase from a lactic acid bacterium, Leuconostoc mesenteroides. Amino Acids 54, 787–798 (2022). https://doi.org/10.1007/s00726-022-03133-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-022-03133-9